Basic Usage
modkit is a bioinformatics tool for working with modified bases from Oxford Nanopore.
![]()
Installation
Pre-compiled binaries are provided for Linux from the release
page. We recommend the use of these in
most circumstances. As a rust-based project, modkit can also be installed with
cargo.
git clone https://github.com/nanoporetech/modkit.git
cd modkit
cargo install --path modkit
# or
cargo install --git https://github.com/nanoporetech/modkit.git
Common Use Cases
- Creating a bedMethyl table with
pileup - Summarizing a modBAM with
modbam summary - Extracting per-read base modification data into a table
- Checking modified base tags in a modBAM
- Making a motif BED file with
motif-bed - Performing differential methylation scoring with
dmr - Investigating differential methylation in direct RNA
- Convert bedMethyl files to bigWig for visualization
- Predict regions of open chromatin on MTase-treated DNA
- Updating and Adjusting MM tags with
adjust-modsandupdate-tags - Convert modification probabilities into hard calls
- Removing base modification calls at the ends of reads
- Narrow analysis to only specific positions with a BED file
- Repairing/adding MM/ML tags to reads with clipped sequences
- Creating hemi-methylation pattern bedMethyl tables with
pileup-hemi
Notes and troubleshooting
- General troubleshooting
- Threshold evaluation examples (for advanced users)
- Querying the logs in
motif search
Constructing bedMethyl tables.
A primary use of modkit is to create summary counts of modified and unmodified bases in an extended bedMethyl format.
bedMethyl files tabulate the counts of base modifications from every sequencing read over each aligned reference genomic position.
In order to create a bedMethyl table, your modBAM must be aligned to a reference genome or transcriptome.
Although not required, providing a reference and using the --modified-bases option will provide the clearest results with the best computational performance.
Only primary alignments are used in generating the table, it is recommended to mark duplicate alignments before running as multiple primary alignments can be double counted.
Recommended usage
Important
Changes for v0.6.0+
For best performance use the --modified-bases option with the base modifications you intend to analyze.
For example:
modkit pileup \
path/to/reads.bam \
path/to/output.bed.gz \
--modified-bases 5mC 5hmC \
--reference path/to/reference.fasta \
--log path/to/log.txt \ # optional, recommended
--bgzf \ # optional
Tip
Note when using a transcriptome-aligned modBAM (for direct RNA), pass the
--preload-referencesflag to increase performance.
Passing --modified-bases not required, but directs Modkit to use optimized routines which will lead to better efficiency.
This option does require a FASTA reference, and will only emit bedmethyl records for base modifications to the primary sequence base in the reference.
For example, a command with the option --modified-bases 5mC 5hmC 6mA will not have 6mA records on reference Cytosine locations where a read has a C>A mismatch nor 5mC/5hmC records at reference Adenine locations.
For more details on how this option changes results from previous versions see migrating to v0.6.0+.
A subset of the base modifications present in the modBAM can specified
For example, passing the option --modified-bases 5mC when the modBAM contains 5mC and 5hmC calls will only produce 5mC records.
Note
If the reference FASTA does not have an index at
path/to/reference.fasta.faione will be created.
Syntax of --modified-bases
You may pass the “long name” such as “5mC” or a primary base and the “short name” separated by a :.
For example, --modified-bases 5mC 5hmC 6mA and --modified-bases C:m C:h A:a are equivalent.
ChEBI codes can also be used.
For example, a to make a pileup for a modBAM with direct RNA reads you may use: --modified-bases A:17596 A:69426 A:a C:m C:19228 G:19229.
Tip
If you don’t know which modified bases are present in your modBAM run:
modkit modbam check-tags $bam --head 100.
Important
If you have a modBAM with very high sequencing depth (>60,000X) use the
--high-depthargument. See limitations for details.
Running without a reference
A reference and --modified-bases is not required, for example:
modkit pileup path/to/reads.bam output/path/pileup.bed --log-filepath pileup.log
This command will produce a bedMethyl record for every position in the reference for which there is at least one base modification call (either modified or unmodified).
This invocation may be slower than using --modified-bases with a reference.
In both cases, a single file (described below) with base count summaries will be created.
The program performs best-practices filtering and manipulation of the raw data stored in the input file. For further details see filtering modified-base calls.
Common options that change how base modifications are tabulated
Narrowing output to CpG dinucleotides
For user convenience, the counting process can be modulated using several additional transforms and filters.
The most basic of these is to report only counts from reference CpG dinucleotides, this behavior is enabled by passing the --cpg flag.
Any motif can be used see below.
modkit pileup path/to/reads.bam output/path/pileup.bed \
--cpg \
--modified-bases 5mC 5hmC \
--ref path/to/reference.fasta
Specifying --combine-mods and/or --combine-strands to simplify output
With palindromic motifs, base modification counts can be summed across strands with --combine-strands:
modkit pileup \
path/to/reads.bam \
output/path/pileup.bed \
--modified-bases 5mC 5hmC \
--cpg \
--combine-strands \
--ref path/to/reference.fasta \
Note that the strand field of the output will be marked as ‘.’ indicating that the strand information has been lost.
Finally, --combine-mods can be used to produce a bedMethyl with binary “modified/not-modified” counts and percentages:
modkit pileup \
path/to/reads.bam \
output/path/pileup.bed \
--modified-bases 5mC 5hmC \
--combine-mods \
--ref path/to/reference.fasta \
[--cpg] \ # optional
[--combine-strands] \ #optional
As can be seen in the above example, these flags can be combined to generate the following output types (flags in [braces] indicate that it is optional):
- Nothing: All modifications at each, stranded, position.
--cpg: Narrow output to only CpGs.--combine-mods [--cpg]: sum counts of all modification types together.--combine-strands --cpg [--combine-mods]: Sum (+)-strand and (-)-strand counts onto the (+)-strand position.
To restrict output to only certain CpGs, pass the --include-bed option with the CpGs or regions to be used, see this page for more details.
modkit pileup path/to/reads.bam output/path/pileup.bed \
--modified-bases 5mC 5hmC \
--cpg \
--ref path/to/reference.fasta \
--include-bed path/to/my_cpgs.bed
Narrowing output to specific motifs
By default, modkit will output a BED row for all genomic positions where there is at least one base modification for the reference base in the input modBAM.
We define a motif as a short DNA sequence potentially containing degenerate codes.
To ease downstream analysis, the --motif <Motif> <offset, 0-based> option can be used to pre-filter and annotate the bedMethyl rows.
The --cpg flag is a alias for --motif CG 0 where the sequence motif is CG and the offset is 0, meaning pileup base modification counts for the first C in the motif on the top strand the second C (complement to G) on the bottom strand.
Another example may be --motif GATC 1, signaling to pileup counts for the A in the second position on the top strand and the A in the third position on the bottom strand.
When multiple motifs are specified the name column (column 4), will indicate which motif the counts are tabulated for.
For example, if --motif CGCG 2 --motif CG 0 are passed you may see lines such as:
oligo_741_adapters 39 40 m,CG,0 4 - 39 40 255,0,0 4 100.00 4 0 0 0 0 0 0
oligo_741_adapters 39 40 m,CGCG,2 4 - 39 40 255,0,0 4 100.00 4 0 0 0 0 0 0
The --combine-strands flag can be combined with --motif however all motifs must be reverse-complement palindromic (CG is a palindrome but CHH is not).
Only one motif at a time is supported with --combine-strands is used (see limitations for details).
Partitioning reads based on phasing information with --phased
If you have a modBAM with phased reads containing a HP tag. These can be partitioned into separate bedMethyl files on output by passing the --phased flag.
modkit pileup path/to/reads.bam output/directory/ \
--cpg --modified-bases 5mC 5hmC --ref <reference.fasta> --phased
The output will be 3 files: hp1.bedmethyl, hp2.bedmethyl, and combined.bedmethyl.
hp1.bedmethyl and hp2.bedmethyl contain counts for records with HP=1 and HP=2 tags, respectively. combined.bedmethyl contains counts for all modBAM records.
For more information on the individual options see the Advanced Usage help document.
Description of bedMethyl output.
Below is a description of the bedMethyl columns generated by modkit pileup. A brief description of the
bedMethyl specification can be found on Encode.
Definitions:
- Nmod - Number of calls passing filters that were classified as a residue with a specified base modification.
- Ncanonical - Number of calls passing filters were classified as the canonical base rather than modified. The
exact base must be inferred by the modification code. For example, if the modification code is
m(5mC) then the canonical base is cytosine. If the modification code isa, the canonical base is adenine. - Nother mod - Number of calls passing filters that were classified as modified, but where the modification is different from the listed base (and the corresponding canonical base is equal). For example, for a given cytosine there may be 3 reads with
hcalls, 1 with a canonical call, and 2 withmcalls. In the bedMethyl row forhNother_mod would be 2. In themrow Nother_mod would be 3. - Nvalid_cov - the valid coverage. Nvalid_cov = Nmod + Nother_mod + Ncanonical, also used as the
scorein the bedMethyl - Ndiff - Number of reads with a base other than the canonical base for this modification. For example, in a row
for
hthe canonical base is cytosine, if there are 2 reads with C->A substitutions, Ndiff will be 2. - Ndelete - Number of reads with a deletion at this reference position
- Nfail - Number of calls where the probability of the call was below the threshold. The threshold can be set on the command line or computed from the data (usually failing the lowest 10th percentile of calls).
- Nnocall - Number of reads aligned to this reference position, with the correct canonical base, but without a base modification call. This can happen, for example, if the model requires a CpG dinucleotide and the read has a CG->CH substitution such that no modification call was produced by the basecaller.
bedMethyl column descriptions.
| column | name | description | type |
|---|---|---|---|
| 1 | chrom | name of reference sequence from BAM header | str |
| 2 | start position | 0-based start position | int |
| 3 | end position | 0-based exclusive end position | int |
| 4 | modified base code and motif | single letter code for modified base and motif when more than one motif is used | str |
| 5 | score | equal to Nvalid_cov | int |
| 6 | strand | ‘+’ for positive strand ‘-’ for negative strand, ‘.’ when strands are combined | str |
| 7 | start position | included for compatibility | int |
| 8 | end position | included for compatibility | int |
| 9 | color | included for compatibility, always 255,0,0 | str |
| 10 | Nvalid_cov | see definitions above. | int |
| 11 | percent modified | (Nmod / Nvalid_cov) * 100 | float |
| 12 | Nmod | see definitions above | int |
| 13 | Ncanonical | see definitions above | int |
| 14 | Nother_mod | see definitions above | int |
| 15 | Ndelete | see definitions above | int |
| 16 | Nfail | see definitions above | int |
| 17 | Ndiff | see definitions above | int |
| 18 | Nnocall | see definitions above | int |
bedRMod output
Important
Changes for v0.6.1
The bedRMod specification includes multiple header lines which all start with the # character.
Enabling this output can be done by passing --bedrmod on the command line, keep in mind that this flag requires --modified-bases and --ref.
Although this output specification is intended for direct RNA base modifications, some of the header fields may be generally useful.
The output table below the header is the same with the exception that column 10 (Nvalid_cov), is substituted with total coverage.
Total coverage is calculated as Nvalid_cov + Nfail.
The score column remains the same: Nvalid_cov.
| column | name | description | type |
|---|---|---|---|
| 10 | Total coverage | Nvalid_cov + Nfail | int |
More details about bedRMod can be found here.
Performance considerations
Important
Changes for v0.6.0
As of version 0.6.0 the efficiency of pileup has been greatly improved.
As a result, fewer threads are often required to get high throughput.
For example, increasing threads beyond 8 a 40X human genome BAM will often not yield much benefit.
Migrating to Modkit v0.6.0+ pileup.
Addition of --modified-bases is the biggest change in Modkit v0.6.0+.
The option --modified-bases 5mC 5hmC or --modified-bases 6mA is roughly equivalent to using --motif C 0 or --motif A 0, respectively, to capture base modification calls only a reference bases for which you are interested in their modification status.
Base modifications at multiple primary bases can be emitted by adding more options to the command line, such as --modified-bases 5mC 5hmC 6mA.
Removal of --preset traditional
Modkit versions v0.6.0 and v0.6.1 do not have the --preset traditional option.
To produce output that is comparable to technologies where 5hmC will be reported as 5mC using best practices provide the following options:
$ modkit pileup $bam $bed --ref $ref
--modified-bases 5mC 5hmC \ # or --modified-bases C
--combine-mods \
--cpg \
--combine-strands
Keep in mind that this output is not exactly equivalent to what --preset traditional would produce previously, see point (1) below for more details.
Changes to pileup algorithm
--ignoreoption has been removed. This option was often a source of confusion and has been removed in favor of the following alternatives:
- Use
--modified-basesto emit records for only the base modifications you’re interested in. For example, a direct RNA run may have many base modifications on each of the primary sequence bases. If you’re only interested in m6A use--modified-bases m6Aand only get records with this base modification. - Use
--combine-modswhen you want to know if a base is “modified vs unmodified”. This can be necessary when comparing to results where all base modifications may be marked as a single base modification.
- Use
--phasedinstead of--partition-tags. The--phasedflag is a more performant version of--partition-tags HP. Instead of an “ungrouped” bedMethyl table, thecombined.bedmethyl[.gz]table contains counts tabulated from all records. - The
--bedgraphflag has been removed. The recommended way to create a “trace”-like output file is to make a bedMethyl and either pipe it directly intomodkit bedmethyl tobigwigor run thetobigwigcommand following bedMethyl creation. - Duplicate read names are no longer logged.
--max-depthremoved. Modkit v0.6.0 has a new algorithm that doesn’t usemax-depth. If you have a modBAM with very high depth and you don’t want to tabulate counts for this depth it is currently recommended to subsample the modBAM before using Modkit.--edge-filter-ed base modification counts are no longer tabulated in Nnocall, they will be completely ignored from tabulation. If this breaks or changes your consumption of these data, please open a GitHub issue.--modified-basesemploys a simpler threshold evaluation algorithm. Prior to v0.6.0, the threshold evaluation was to take the most likely passing base modification call. This lead to confusion about how to “tune” this parameter to tweak results. When using--modified-bases(strongly recommended) the algorithm is now simply to use the most likely call (the one with the highest probability) and determine if it is pass or fail by whether or not it is less than the threshold for that base or modification. In almost all cases this simpler algorithm produces the same results and is what the user expected in the first place.
Limitations
- As of v.0.6.0
--modified-basesdoes not support duplex base modification calls. To enable usage of base modification calls on duplex reads, pass the--duplexflag. This limitation will be removed in the future. --combine-strandswhen using multiple palindromic motifs is no longer implemented. If this is a crucial step in your workflow, please open a GitHub issue. The new workflow is to run with a single motif and--combine-strandsfor each motif. Modkit v0.6.0 is substantially quicker and lighter on resource requirements such that running multiple times will likely be quicker than the older versions.--invert-edge-filterremoved. This option is still available inmodkit modbam adjust-mods.- In Modkit versions v0.6.0 and v0.6.1
pileupwill saturate depth at 65,535 (maximum for a unsigned 16-bit integer). If your modBAM has a depth greater than this value, it is recommended to use the--high-depthflag so that 65,535 reads will be used at each genomic position. To achieve greater depth, divide the modBAM into subsets with depth <= 65,535, runpileupon each, thenmodbam merge. This limitation will be removed in the future when it can be proven not to have any performance or resource regression.
Make hemi-methylation bedMethyl tables with pileup-hemi
Base modifications in DNA are inherently single-stranded, they (usually [^1]) don’t change the base pairing of the modified base. However, it may be of interest to know the correspondence between the methylation state of a single base and another nearby base on the opposite strand - on the same molecule. In CpG dinucleotides, this is called “hemi-methylation”, when one cytosine is methylated and the neighbor on the opposite strand is not:
m
5'GATCGTACA
CTAGCATGT
-
In the above diagram, the cytosine in the fourth position on the positive strand is methylated (5mC) and the cytosine in the fifth position is canonical (-), indicating a “hemi-methylation”.
In the case of 5mC and canonical, there are 4 “patterns” of methylation:
m,m (5mC, 5mC)
-,m (canonical, 5mC)
m,- (5mC, canonical)
-,- (canonical, canonical)
These are all measured at the single molecule level, meaning each molecule must report on both strands (as
is the case with duplex reads). For CpGs in the example above the
MM tags would be C+m? and G-m? for the top-strand and bottom-strand cytosines, respectively.
The modkit pileup-hemi command will perform an aggregation of the methylation “patterns” at genomic positions. An example
command to perform hemi-methylation analysis at CpGs would be
modkit pileup-hemi \
/path/to/duplex_reads.bam \
--cpg \
-r /path/to/reference.fasta \
-o hemi_pileup.bed \
--log modkit.log
Many of the pileup options are available in pileup-hemi with a couple differences: :
- A motif must be provided. The
--cpgflag is a preset to aggregate CpG hemi-methylation patterns as shown above. If a motif is provided (as an argument to--motif) it must be reverse-complement palindromic. - A reference must be provided.
- Both the positive strand base modification probability and the negative strand base modification probability must be above the pass threshold.
See Advanced Usage for details on all the options.
Description of hemi-methylation patterns
The modkit pileup-hemi command aggregates a pair of base modification calls at each reference motif position
for each double-stranded DNA molecule. The base modification “pattern” indicates the methylation state on each base
in 5-prime to 3-prime order, using the base modification code to indicate the identity of the base modification and
- to indicate canonical (unmodified). For example m,-,C would mean the first base (from the reference 5’ direction)
is 5mC and the second base is unmodified and the primary base is cytosone. Similarly, h,m,C indicates the first base is
5hmC and the second base is 5mC. The primary base called by the read is included to help disambiguate the unmodified
patterns (-,-). All patterns recognized at a location will be reported in the bedMethyl output.
Definitions:
- Npattern - Number of call-pairs passing filters that had the pattern and primary base in column 4. E.g.
m,-,Cindicates the first base in the 5’ to 3’ direction is 5mC, the second base is unmodified and the primary base in the reads was C. - Ncanonical - Number of call-pairs passing filters that were classified as unmodified (i.e. the pattern is
-,-). - Nother_pattern - Number of call-pairs passing filters where the pattern is different from the pattern in
column 4, but where the primary read base is the same. This count includes the unmodified pattern (
-,-). Note this differs frompileupwhere Nother does not contain the canonical counts. - Nvalid_cov - the valid coverage, total number of valid call-pairs.
- Ndiff - Number of reads with a primary base other than the primary base in column 4.
- Ndelete - Number of reads with a deletion at this reference position.
- Nfail - Number of call-pairs where the probability of the at least one of the calls in the pair was below the pass threshold. The threshold can be set on the command line or computed from the data (usually failing the lowest 10th percentile of calls).
- Nnocall - Number of reads where either one or both of the base modification calls was not present in the read.
bedMethyl column descriptions.
| column | name | description | type |
|---|---|---|---|
| 1 | chrom | name of reference sequence from BAM header | str |
| 2 | start position | 0-based start position | int |
| 3 | end position | 0-based exclusive end position | int |
| 4 | methylation pattern | comma-separated pair of modification codes - means canonical, followed by the primary read base | str |
| 5 | score | equal to Nvalid_cov | int |
| 6 | strand | always ‘.’ because strand information is combined | str |
| 7 | start position | included for compatibility | int |
| 8 | end position | included for compatibility | int |
| 9 | color | included for compatibility, always 255,0,0 | str |
| 10 | Nvalid_cov | see definitions above | int |
| 11 | fraction modified | Npattern / Nvalid_cov | float |
| 12 | Npattern | see definitions above | int |
| 13 | Ncanonical | see definitions above | int |
| 14 | Nother_pattern | see definitions above | int |
| 15 | Ndelete | see definitions above | int |
| 16 | Nfail | see definitions above | int |
| 17 | Ndiff | see definitions above | int |
| 18 | Nnocall | see definitions above | int |
Limitations
- Only one motif can be used at a time, this limitation may be removed in a later version.
- Partitioning on tag key:value pairs is not currently supported.
[^1] In biology, there are almost always exceptions to every rule!
Updating and Adjusting MM tags.
The adjust-mods subcommand can be used to manipulate MM (and corresponding ML) tags in a
modBam. In general, these simple commands are run prior to pileup, visualization, or
other analysis. For adjust-mods and update-tags, if a correct MN tag is found, secondary and supplementary
alignments will be output. See troubleshooting for details.
Ignoring a modification class.
To remove a base modification class from a modBAM and produce a new modBAM, use the
--ignore option for adjust-mods.
modkit modbam adjust-mods input.bam output.adjust.bam --ignore <mod_code_to_ignore>
For example the command below will remove 5hmC calls, leaving just 5mC calls.
modkit modbam adjust-mods input.bam output.adjust.bam --ignore h
For technical details on the transformation see Removing modification calls from BAMs.
Combining base modification probabilities.
Combining base modification probabilities may be desirable for downstream analysis or
visualization. Unlike --ignore which removes the probability of a class, --convert
will sum the probability of one class with another if the second class already exists. For
example, the command below will convert probabilities associated with h probability into
m probability. If m already exists, the probabilities will be summed. As described in
changing the modification code,
if the second base modification code doesn’t exist, the probabilities are left unchanged.
modkit modbam adjust-mods input.bam output.convert.bam --convert h m
Updating the flag (? and .).
The specification (Section 1.7) allows
for omission of the MM flag, however this may not be the intent of missing base
modification probabilities for some models. The command below will add or change the ? flag to a modBAM.
modkit modbam adjust-mods input.bam output.bam --mode ambiguous
Another option is to set the flag to ., the “implicitly canonical” mode:
modkit modbam adjust-mods input.bam output.bam --mode implicit
Changing the base modification code.
Some functions in modkit or other tools may require the mod-codes in the MM tag be in
the specification.
For example, the following command will change C+Z, tags to C+m, tags.
modkit modbam adjust-mods input.bam output.bam --convert Z m
Filtering to specific motifs
If you want to remove base modification calls that don’t match a specific basecall sequence motif, you can use the --motif in adjust-mods retain only base modification calls that match the motif.
The format for specifying the motif is <sequence> <offset> IUPAC codes are allowed in the motif sequence and <offset> specifies the 0-based offset into the sequence for the primary base carrying the modification.
For example for CpG dinucleotides --motif CG 0.
Inspecting base modification probabilities, modbam sample-probs
Calculate an estimate of the base modification probability distribution and estimate filter thresholds at various percentiles.
For details on how base modification probabilities are calculated, see the FAQ page
For most use cases the automatic filtering enabled in modkit will produce nearly ideal results.
However, in some cases such as exotic organisms or specialized assays, you may want to interrogate the base modification probabilities directly and tune the pass thresholds.
The modkit sample-probs command is designed for this task.
There are two ways to use this command, first by simply running modkit sample-probs $mod_bam to get a tab-separated file of threshold values for each modified base.
This can save time in downstream steps where you wish to re-use the threshold value by passing --filter-threshold and skip re-estimating the value.
To generate more advanced output, add --hist --out-dir $output_dir to the command and generate per-modification histograms of the output probabilities.
Using the command this way produces 3 files in the $output_dir:
- An HTML document containing a histogram of the total counts of each probability emitted for each modification code (including canonical) in the sampled reads.
- Another HTML document containing the proportions of each probability emitted.
- A tab-separated table with the same information as the histograms and the percentile rank of each probability value.
The schema of the table is as follows:
| column | name | description | type |
|---|---|---|---|
| 1 | code | modification code or ‘-’ for canonical | string |
| 2 | primary base | the primary DNA base for which the code applies | string |
| 3 | range_start | the inclusive start probability of the bin | float |
| 4 | range_end | the exclusive end probability of the bin | float |
| 5 | count | the total count of probabilities falling in this bin | int |
| 6 | frac | the fraction of the total calls for this code/primary base in this bin | float |
| 7 | percentile_rank | the percentile rank of this probability bin | float |
From these plots and tables you can decide on a pass threshold per-modification code and use --mod-threshold/--filter-threshold accordingly.
Summarizing a modBAM.
The modkit modbam summary sub-command is intended for collecting read-level or genome-level statistics from a modBAM.
It can optionally sample a fraction of the reads, sample a target number of reads, and subset to a region.
Important
Changes for v0.6.0+ The
--no-samplingoption has been removed, using the entire modBAM is now the default. The default behavior is to gather a summary of the entire modBAM. The fastest run time will be to use--num-reads, followed by--sample-frac.
Summarize the base modification calls in a modBAM.
modkit modbam summary input.bam
will output a table similar to this
> not subsampling, using all reads
> calculating threshold that removes lowest 10 percentile of calls
> collecting base modification calls from unmapped records
# bases C,A
# total_reads_used 8340404
# count_reads_C 8340404
# count_reads_A 8340404
# pass_threshold_A 0.7128906
# pass_threshold_C 0.7089844
# modification_codes_for_A a
# modification_codes_for_C h,m
base code pass_count pass_frac all_count all_frac
A - 38789589231 0.9522225 41493100163 0.91872257
A a 1946256088 0.04777748 3670806258 0.08127743
C - 26729831806 0.95418394 28992497768 0.9339372
C h 113554922 0.0040536085 374670368 0.01206928
C m 1169904115 0.041762467 1676137832 0.053993538`
Important
Changes for v0.6.0+
Summarize only the matched base modification calls
When a reference is passed to modkit modbam summary the program can be set to only record base modifications on reads where the base in the read matches the reference base.
This may sound intuitive, but the alternative is to record base modifications on mismatches, the default (e.g. a 5mC call on a A>C mismatch).
To enable this behavior, pass the --matched-only flag.
When this flag is passed, only mapped records are used.
Another option is to use --motif, for example --motif DRACH 2.
This has the effect of only calculating statistics at certain reference motifs and the base in the read must match.
Description of columns in modkit summary:
Totals table
The lines of the totals table are prefixed with a # character.
| row | name | description | type |
|---|---|---|---|
| 1 | bases | comma-separated list of canonical bases with modification calls. | str |
| 2 | total_reads_used | total number of reads from which base modification calls were extracted | int |
| 3+ | count_reads_{base} | total number of reads that contained base modifications for {base} | int |
| 4+ | modification_codes_for_{base} | comma-separated list of modification codes found for this base | str |
| 5+ | filter_threshold_{base} | filter threshold used for {base} | float |
Modification calls table
The modification calls table follows immediately after the totals table.
| column | name | description | type |
|---|---|---|---|
| 1 | base | canonical base with modification call | char |
| 2 | code | base modification code, or - for canonical | char |
| 3 | pass_count | total number of passing (confidence >= threshold) calls for the modification in column 2 | int |
| 4 | pass_frac | fraction of passing (>= threshold) calls for the modification in column 2 | float |
| 5 | all_count | total number of calls for the modification code in column 2 | int |
| 6 | all_frac | fraction of all calls for the modification in column 2 | float |
For more details on thresholds see filtering base modification calls.
There are --filter-percentile, and --filter-threshold options that
can be used with or without sampling.
Passing a threshold directly.
To estimate the pass thresholds on a subset of reads, but then summarize all of the
reads, there is a two-step process. First, determine the thresholds with modkit modbam sample-probs (see usage for more details). Then run
modkit modbam summary with the threshold value specified.
modkit modbam sample-probs input.bam [--sampling-frac <frac> | --num-reads <num>]
This command will output a table like this:
> sampling 10042 reads from BAM
base percentile threshold
C 10 0.6972656
C 50 0.96484375
C 90 0.9941406
You can then use pass this threshold directly to modkit modbam summary:
modkit modbam summary input.bam --filter-threshold 0.6972656
Sampling the modBAM
Important
Changes for v0.6.0+
Often summarizing the entire modBAM is unnecessary to get a quick answer. There are 3 ways to sub-sample the modBAM:
- Use
--region(with aligned BAM only) to look at only a specific region - Use
--num-readsto sample approximately this many reads from the BAM. See the details on sampling for how this algorithm works. - Use
--sampling-frac(optionally with-seed).
Using --num-reads is often the fastest.
Performance considerations
When using --ignore-index or an unaligned modBAM, memory consumption should be stable and low.
In this case the execution time is bounded by the speed with which the program can read the BAM (set --io-threads) and parse the MM tag.
If you have an sorted, indexed modBAM the work can be divided up into intervals and processed in parallel.
Adding more --threads will generally speed up execution time proportionally, however it will require more memory.
Each worker (“thread”) holds a histogram of probabilities that it uses compiles as it scans over it’s section of the modBAM.
There is always a stable number of these histograms initialized at the start of execution, but there will be more of them initialized with more threads.
Calculating modification statistics in regions
There are many analysis operations available in modkit once you’ve generated a bedMethyl table.
One such operation is to calculate aggregation statistics on specific regions, for example in CpG islands or gene promoters.
The modkit stats command is designed for this purpose.
# these files can be found in the modkit repository
cpgs=tests/resources/cpg_chr20_with_orig_names_selection.bed
sample=tests/resources/lung_00733-m_adjacent-normal_5mc-5hmc_chr20_cpg_pileup.bed.gz
modkit stats ${sample} --regions ${cpgs} -o ./stats.tsv [--mod-codes "h,m"]
Note that the argument
--mod-codescan alternatively be passed multiple times, e.g. this is equivalent:
--mod-codes c --mod-codes h
The output TSV has the following schema:
| column | Name | Description | type |
|---|---|---|---|
| 1 | chrom | name of reference sequence from BAM header | str |
| 2 | start position | 0-based start position | int |
| 3 | end position | 0-based exclusive end position | int |
| 4 | name | name of the region from input BED (. if not provided) | str |
| 5 | strand | Strand (+, -, .) from the input BED (. assumed for when not provided) | str |
| 6+ | count_x | total number of x base modification codes in the region | int |
| 7+ | count_valid_x | total valid calls for the primary base modified by code x | int |
| 8+ | percent_x | count_x / count_valid_x * 100 | float |
Columns 6, 7, and 8 are repeated for each modification code found in the bedMethyl file or provided with --mod-codes argument.
An example output:
chrom start end name strand count_h count_valid_h percent_h count_m count_valid_m percent_m
chr20 9838623 9839213 CpG: 47 . 12 1777 0.6752954 45 1777 2.532358
chr20 10034962 10035266 CpG: 35 . 7 1513 0.46265697 0 1513 0
chr20 10172120 10172545 CpG: 35 . 15 1229 1.2205045 28 1229 2.278275
chr20 10217487 10218336 CpG: 59 . 29 2339 1.2398461 108 2339 4.617358
chr20 10433628 10434345 CpG: 71 . 29 2750 1.0545455 2 2750 0.07272727
chr20 10671925 10674963 CpG: 255 . 43 9461 0.45449743 24 9461 0.25367296
Calling mods in a modBAM
The call-mods subcommand in modkit transforms one modBAM into another
modBAM where the base modification probabilities have been clamped to 100% and
0%. If the --filter-threshold and/or --mod-threshold
options are provided, base modification calls
failing the threshold will be removed prior to changing the probabilities. The
output modBAM can be used for visualization, pileup, or other applications.
For call-mods, if a correct MN tag is found, secondary and supplementary
alignments will be output. See troubleshooting for details.
A modBAM that has been transformed with call-mods using --filter-threshold
and/or --mod-threshold cannot be re-transformed with different thresholds.
Note on pileup with clamped probabilities: modkit pileup will attempt to
estimate the threshold probability by default, but it is unnecessary if the
modBAM is the result of call-mods. The threshold probabilities will be
artificially high (i.e. not representative of the model’s output
probabilities). Similarly, specifying --filter-threshold and
--mod-threshold is not useful because all the ML probabilities have been set
to 0 and 100%.
Example usages
Estimate the threshold on the fly, apply to modBAM and clamp the modification calls to certainty.
modkit modbam call-mods <in.bam> <out.bam>
Specify a filter threshold for your use-case
modkit modbam call-mods <in.bam> <out.bam> --filter-threshold A:0.9 --mod-threshold a:0.95 --filter-threshold C:0.97
Call mods with the estimated threshold and ignore modification calls within 100 base pairs of the ends of the reds
modkit modbam call-mods <in.bam> <out.bam> --edge-filter 100
Removing modification calls at the ends of reads
If you have reads where you know base modifications near the ends should not be used
(for example, if they are in adapters), you can use the --edge-filter <n_basepairs> option.
Two comma-separated values may be provided to asymmetrically filter out
base modification calls from the start and end of reads. For example, 4,8 will
filter out base modification calls in the first 4 and last 8 bases of the read. One value
will filter symmetrically.
pileup, will ignore base modification calls that are<n_basepairs>from the ends.adjust-mods, will remove base modification calls that are<n_basepairs>from the ends from the resultant output modBAM.summary, will ignore base modification calls that are<n_basepairs>from the ends.sample-probs, will ignore base modification calls that are<n_basepairs>from the ends.call-mods, will remove base modification calls that are<n_basepairs>from the ends from the resultant output modBAM.extract, will ignore base modification calls that are<n_basepairs>from the ends, this also applies when making the read-calls table (see intro to extract).
In pileup, call-mods, and extract the edge-filter is also respected when estimating the pass-thresholds.
All commands have the flag --invert-edge-filter that will keep only base modification probabilities within
<n_basepairs> of the ends of the reads.
Example usages
Call mods with the estimated threshold and ignore modification calls within 100 base pairs of the ends of the reads
modkit call-mods <in.bam> <out.bam> --edge-filter 100
Perform pileup, ignoring base modification calls within 100 base pairs of the ends of the reads
modkit pileup <in.bam> <out.bed> --edge-filter 100
Filter out base modification calls within the first 25 bases or the last 10 bases.
modkit pileup <in.bam> <out.bed> --edge-filter 25,10
Repair MM/ML tags on trimmed reads
The modkit repair command is useful when you have a BAM with reads where the
canonical sequences have been altered in some way that either renders the MM
and ML tags invalid (for example, trimmed or hard-clipped) or the data has
been lost completely. This command requires that you have the original base
modification calls for each read you want to repair, and it will project these
base modification calls onto the sequences in the altered BAM.
The command uses two arguments called the “donor” and the “acceptor”. The
donor, contains the original, correct, MM and ML tags and the acceptor is
either missing MM and ML tags or they are invalid (they will be discarded
either way). The reads in the donor must be a superset of the reads in the
acceptor, meaning you can have extra reads in the donor BAM if some reads have
been removed or filtered earlier in the workflow. Both the donor and the
acceptor must be sorted by read name prior to running modkit repair.
Duplicate reads in the acceptor are allowed so long as they have valid SEQ
fields. Lastly, modkit repair only works on reads that have been trimmed,
other kinds of alteration such as run-length-encoding are not currently
supported. Split reads, or other derived transformations, are not currently
repairable with this command.
For example a typical workflow may look like this:
# original base modification calls
basecalls_5mC_5hmC.bam
# basecalls that have been trimmed
trimmed.bam # could also be fastq, but would require conversion to BAM
# the two BAM files need to be sorted
samtools -n trimmed.bam -O BAM > trimed_read_sort.bam
samtools -n basecalls_5mC_5hmC.bam -O BAM > basecalls_5mC_5hmC_read_sort.bam
modkit modbam repair \
--donor-bam basecalls_5mC_5hmC_read_sort.bam \
--acceptor-bam trimed_read_sort.bam \
--log-filepath modkit_repair.log \
--output-bam trimmed_repaired.bam
Working with sequence motifs
The modkit motif suite contains tools for discovery and exploration of short degenerate sequences (motifs) that may be enriched in a sample.
A common use case is to discover the motifs enriched for modification in a native bacterial sample which can give indication of methyltransferase enzymes present in the genomes present in the sample.
The following tools are available:
- Find enriched motifs de novo from a bedMethyl with
search. evaluateorrefinea table of known motifs- Making a motif BED file with
motif bed
Making a motif BED file.
Downstream analysis may require a BED file to select motifs of interest. For example, selecting GATC motifs in E. coli. This command requires a reference sequence in FASTA a motif to find, which can include IUPAC ambiguous bases and a position within the motif.
The following command would make a BED file for CG motifs.
modkit motif-bed reference.fasta CG 0 1> cg_modifs.bed
The output is directed to standard out.
Find highly modified motif sequences
The modkit find-motifs command will attempt to summarize short genome sequences (motifs) that are more found to be highly modified (i.e. enriched for methylation).
The input to this command is a bedMethyl generated by modkit pileup and the reference sequence used.
For example, to run the command with default settings (recommended):
bedmethyl=/path/to/pileup.bed
ref=/path/to/reference.fasta
modkit motif search \
-i ${bedmethyl} \
-r ${ref} \
-o ./motifs.tsv \
--threads 32 \
--log ./modkit_find_motifs_log.txt
Specifying an output with -o will generate a machine-readable tab-separated-values file, a human-readable version of the table will always be logged to the terminal and the logfile.
If you have compressed your bedMethyl table with bgzip you can use the compressed table as well.
A compressed table with tabix-generated index is required to use the --contig option which will find motifs using only a single contig in the bedMethyl table.
This may be useful for applications such as metagenomics.
Output format
All output tables are output in two formats, machine-readable and human-readable. The human-readable tables are always output to the log and terminal, the machine-readable tables are output to files specified on the command line.
Machine-readable table
| column | name | description | type |
|---|---|---|---|
| 1 | mod_code | code specifying the modification found in the motif | str |
| 2 | motif | sequence of identified motif using IUPAC codes | str |
| 3 | offset | 0-based offset into the motif sequence of the modified base | int |
| 4 | frac_mod | fraction of time this sequence is found in the high modified set col-5 / (col-5 + col-6) | float |
| 5 | high_count | number of occurances of this sequence in the high-modified set | int |
| 6 | low_count | number of occurances of this sequence in the low-modified set | int |
| 7 | mid_count | number of occurances of this sequence in the mid-modified set | int |
Human-readable table
| column | name | description | type |
|---|---|---|---|
| 1 | motif | human-readable representation of the motif sequence with the modification code in brackets | str |
| 2 | frac_mod | fraction of time this sequence is found in the high modified set col-3 / (col-3 + col-4) | float |
| 3 | high_count | number of occurances of this sequence in the high-modified set | int |
| 4 | low_count | number of occurances of this sequence in the low-modified set | int |
| 5 | mid_count | number of occurances of this sequence in the mid-modified set | int |
Specifying known motifs
Multiple motif sequences suspected to be present can be specified with the --known-motif option.
A machine-readable table of the motif sequences that are not found during the search can be specified with the --known-motifs-table option.
Using this option will add two columns to the above tables:
| name | description | type |
|---|---|---|
| status | equal, subset, superset, intersect, or disjoint describes the relationship of the discovered motif to the known motif, more details below | str |
| closest_known_motif | of all motifs specified with --known-motif the one that is most similar to the discovered motif | str |
The status column is the relationship of the set of sequences described by the motifs.
Say you have two motifs A and B they represent a set of sequences \(\mathbf{A}\) and \(\mathbf{B}\) .
For example, the motif [a] represents all sequences with at least one A primary base, whereas the set of sequences represented by G[a]TC is only {GATC}.
The status fills in the blank in the statement: \(\mathbf{A}\) \(?\) \(\mathbf{B}\).
If any of the known motifs are not found during the search process an additional table is also emitted in machine- and human-readable versions.
Machine-readable table
| column | name | description | type |
|---|---|---|---|
| 1 | mod_code | code specifying the modification found in the motif | str |
| 2 | motif | sequence of identified motif using IUPAC codes | str |
| 3 | offset | 0-based offset into the motif sequence of the modified base | int |
| 4 | frac_mod | fraction of time this sequence is found in the high modified set col-5 / (col-5 + col-6) | float |
| 5 | high_count | number of occurances of this sequence in the high-modified set | int |
| 6 | low_count | number of occurances of this sequence in the low-modified set | int |
| 7 | mid_count | number of occurances of this sequence in the mid-modified set | int |
| 8 | status | equal, Subset, Superset, or Disjoint describes the relationship of the known motif to the closest discovered motif | str |
| 9 | closest_found_motif | which of the discovered motifs is most simuilar to the known motif | str |
Human-readable table
| column | name | description | type |
|---|---|---|---|
| 1 | motif | human-readable representation of the motif sequence with the modification code in brackets | str |
| 2 | frac_mod | fraction of time this sequence is found in the high modified set col-3 / (col-3 + col-4) | float |
| 3 | high_count | number of occurances of this sequence in the high-modified set | int |
| 4 | low_count | number of occurances of this sequence in the low-modified set | int |
| 5 | mid_count | number of occurances of this sequence in the mid-modified set | int |
| 6 | status | equal, Subset, Superset, or Disjoint describes the relationship of the known motif to the closest discovered motif | str |
| 7 | closest_found_motif | which of the discovered motifs is most simuilar to the known motif | str |
Simple description of the search algorithm
The first step in find-motifs is to categorize each genomic position in the pileup into one of three groups based on the fraction modified column in the bedMethyl:
- Low-modified
- Mid-modified
- High-modified
The threshold values for each group can be set on the command line (--high-thresh and --low-thresh).
For example, consider a high threshold of 0.6 and a low threshold of 0.2 the following 3 bedMethyl records would be put into the high-, low-, and mid-groups, respectively:
contig1 6 7 a 27 - 6 7 255,0,0 27 96.30 26 1 0 0 3 0 0
contig1 8 9 a 24 - 8 9 255,0,0 24 4.17 1 23 0 0 5 1 0
contig1 218 219 a 21 + 218 219 255,0,0 21 28.57 6 15 0 2 3 0 0
The sequence around each modified position is then collected from the reference FASTA file.
The length of the sequence can be set with the --context-size option, accepting two values: the number of bases upstream and the number of bases downstream of the modified location.
For example --context-size 12 12 will collect 12 bases upstream and 12 bases downstream of the modified base for a maximum motif length of 25 base pairs.
The algorithm then iteratively expands, contracts, and merges sequences while the following criteria are met:
- The number of occurrences in the high-modified set is greater than
min_sites(set by--min-sites). - The fraction \( \frac{\textit{H}}{\textit{H} + \textit{L}} \), is greater than
frac_mod(set with--min-frac-mod), where \( \textit{H} \) and \( \textit{L} \) is the number of total sequence contexts in the high-modified and low-modified set, respectively. - The log-odds of the context being in the high-modified category is greater than
min_log_odds(set with--min-log-odds).
Once a motif sequence cannot be changed (made more general or more restrictive) without violating one of these criteria, the motif sequence is considered complete. As the algorithm continues, context sequences that match discovered sequences are removed from consideration.
A secondary search step, called the “exhaustive search”, is also performed by starting with every k-mer (where k is less than the total sequence length, 3 by default, set with --exhaustive-seed-len) at every motif position.
The log-odds threshold for this search is usually higher and set with --exhaustive-seed-min-log-odds.
Decreasing this value can drastically increase computational time, see the next section for more details.
Options to decrease or limit search time.
The optional (but recommended) exhaustive search step checks many sparsely defined sequences, called seeds, for enrichment as defined by --exhaustive-seed-min-log-odds.
The algorithm then refines the seeds above the threshold into candidate motifs using the method described previously.
An example seed is GNNNNNNNNNNC[m]NNNNNNNNNNNA using the modification code for 5mC.
The number of seeds is defined by the --context-size and --exhaustive-seed-length parameters.
For example, if --context-size a b is passed the seeds will be length \( a + b + 1 \).
The default value is a = 12, b = 12 so the sequence length is 25, but we only use 24 of those positions, call this \( n \).
The default value for --exhaustive-seed-length is 3, call this \( k \).
Remember that there are 4 primary sequence bases.
So the number of seeds to search is \( k^4 * \binom{n}{k} \) or 129,536 seeds.
Most of these seeds will fall below the log-odds required, and not follow-on to refinement.
However, if a large number ( \( \gt 500 \) ) are above the log-odds threshold then this step can be very time consuming.
There are a couple options that can decrease or limit this run time, whist usually affording identical or very good results.
- A simple timeout. When the search starts, the top-N (as ranked by log-odds) seeds are taken. The search is run on this “batch”, if the timeout has not expired the next batch of N is taken. If all of the seeds are not evaluated before running out of time, the results are returned but an ERROR message is displayed.
# specify that search should stop after 15 minutes
$ modkit motif search ... --search-timeout 15m
- Top percent.
Only search the top P-% of seeds (or a maximum number).
So a command like this
--search-top-pct 20 --max-exhaustive-seeds 200would search the top 20% of seeds as ranked by their log-odds for being motifs or the top 200 seeds if 20% of all of the seeds passing--exhaustive-seed-min-log-oddsis greater than 200. n.b. With default settings, in the most pathological settings 129,536 could be searched.
# only search the min(top 20% of seeds, 100 seeds)
$ modkit motif search ... --search-top-pct 20 --max-exhaustive-seeds 100
- Batch and Narrow Optimization. This algorithm is a combination of (1) and (2). In this scenario, we take the top P-% of seeds (or the maximum allowed) as in (2). But instead of stopping as in (2) we continue (as in (1)) but we remove contexts from consideration based on the motifs we’ve found before the next round. At the end of the round, if zero motifs are found, we stop.
# same as (2), but continue to search after each batch
$ modkit motif search .. --search-top-pct 20 --max-exhaustive-seeds 100 --narrow-search
- Batch and Narrow Optimization with Timeout. This scenario is the same as (3), but if we’ve run out of time at the end of a round - stop.
# same as (3), but continue to search after each batch, until we've spent 15 minutes searching.
$ modkit motif search .. --search-top-pct 20 --max-exhaustive-seeds 100 --narrow-search --search-timeout 15m
Tuning parameters and --skip-search
The default parameters have been picked to be sufficiently sensitive, however if you decide to adjust the parameters in general increasing sensitivity will increase compute time.
- Increasing the
--min-frac-modwill stop search earlier which will decrease compute time. - Decreasing
--min-siteshas the largest effect and can especially cause the secondary search to crawl more sequences. Decreasing--min-sitesalong with--skip-searchmay be a useful technique to find very rare sequence motifs. - Increasing
--exhaustive-seed-min-log-oddscan drastically decrease compute time (sometimes while maintaining sensitivity).
Also consider the additional steps in performance considerations.
Investigating how motifs are evaluated
The motif search function uses JSON-lines structured logging that can be searched and queried for specific events.
The schema and some examples are described in another section.
Evaluate a table of known motifs
The modkit search command has an option to provide any number of known motifs with --know-motif.
However, if you already have a list of candidate motifs (e.g. from a previous run of modkit motif search) you can check these motifs quickly against a bedMethyl table with modkit motif evaluate.
modkit motif evaluate -i ${bedmethyl} --known-motifs-table motifs.tsv -r ${ref}
Similarly, the search algorithm can be run using known motifs as seeds:
modkit motif refine -i ${bedmethyl} --known-motifs-table motifs.tsv -r ${ref}
The output tables to both of these commands have the same schema:
| column | name | description | type |
|---|---|---|---|
| 1 | mod_code | code specifying the modification found in the motif | str |
| 2 | motif | sequence of identified motif using IUPAC codes | str |
| 3 | offset | 0-based offset into the motif sequence of the modified base | int |
| 4 | frac_mod | fraction of time this sequence is found in the high modified set col-5 / (col-5 + col-6) | float |
| 5 | high_count | number of occurances of this sequence in the high-modified set | int |
| 6 | low_count | number of occurances of this sequence in the low-modified set | int |
| 7 | mid_count | number of occurances of this sequence in the mid-modified set | int |
| 8 | log_odds | log2 odds of the motif being in the high-modified set | int |
In the human-readable table columns (1) and (2) are merged to show the modification code in the motif sequence context, the rest of the columns are the same as the machine-readable table.
Structured logs in modkit motif search
The debug logs can me emitted using the --log-filepath option to modkit motif search.
These logs are JSON-formatted lines that can be queried with a tool such as jq or jaq.
The top-level schema is:
| name | description |
|---|---|
| timestamp | system time of the log message |
| level | log level, ERROR, WARN, INFO, or DEBUG |
| fields | JSON object with more information about the event (details belos) |
| target | Module logging the message (not usually very useful) |
| filename | Filename where the log message originated |
| line_number | Line in the file where the log message originated |
For example, to print out the INFO logs to the terminal:
cat ${log} | jq 'select(.level == "INFO") | .fields.message'
The fields object contains more information that can be useful for drilling down into what happened during a search.
| name | required | description |
|---|---|---|
| message | true | The human-readable log message |
| mod_code | false | The modification code (e.g. a) being worked on when this event was emitted |
| stage | false | The stage of the search algorithm, one of {seeded, seedless, search} |
| motif | false | The motif under consideration, this is the hunam-readable name e.g. G[a]TC |
| from_motif | false | In the refinement step, this is the “input” motif, usually a seed, the same notation as motif is used |
| action | false | One of {found, refined, discard} |
| require | false | Required value (only present when action = discard) |
| value | false | The value this motif has (only present when action = discard) |
Actions
foundA motif passes all criteria, and is kept. (It may later be decided that it was re-found anddiscarded).refinedWhen a motif “seed” is transformed into a motif that passes criteria, usually the step right beforefound, but connects this motif to the seed orfrom_motifdiscardWhen a motif is proposed byrefined, but fails for some reason, such as it was already discovered
Examples:
- Messages for a motif:
cat ${log} | jq 'select(.fields.motif == "TCG[a]") | .fields.message'
- Information for when a motif was refined:
cat ${log} | jq 'select(.fields.from_motif == "R[a]GC") | .fields'
- Check all of the seeds that were refined:
cat ${log} | jq 'select(.fields.stage == "seeded" and .fields.action == "found" and .fields.mod_code == "a") | .fields'
Extracting base modification information
The modkit extract full sub-commands will produce a table containing the base modification probabilities, the read sequence context, and optionally aligned reference information.
For extract full and extract calls, if a correct MN tag is found, secondary and supplementary alignments may be output with the --allow-non-primary flag.
See troubleshooting for details.
The table will by default contain unmapped sections of the read (soft-clipped sections, for example).
To only include mapped bases use the --mapped flag. To only include sites of interest, pass a
BED-formatted file to the --include-bed option. Similarly, to exclude sites, pass a BED-formatted
file to the --exclude option. One caution, the files generated by modkit extract can be large (2-2.5x
the size of the BAM). You may want to either use the --num-reads option, the --region option, or
pre-filter the modBAM ahead of time. You can also stream the output to stdout by setting the output to -
or stdout and filter the columns before writing to disk.
Description of output table for extract full
| column | name | description | type |
|---|---|---|---|
| 1 | read_id | name of the read | str |
| 2 | forward_read_position | 0-based position on the forward-oriented read sequence | int |
| 3 | ref_position | aligned 0-based reference sequence position, -1 means unmapped | int |
| 4 | chrom | name of aligned contig, or ‘.’ if the read is Gunmapped | str |
| 5 | mod_strand | strand of the molecule the base modification is on | str |
| 6 | ref_strand | strand of the reference the read is aligned to, or ‘.’ if unmapped | str |
| 7 | ref_mod_strand | strand of the reference with the base modification, or ‘.’ if unmapped | str |
| 8 | fw_soft_clipped_start | number of bases soft clipped from the start of the forward-oriented read | int |
| 9 | fw_soft_clipped_end | number of bases soft clipped from the end of the forward-oriented read | int |
| 10 | alignment_start | leftmost (i.e. smallest) aligned reference position | int |
| 11 | alignment_end | rightmost (i.e. largest) aligned reference position | int |
| 12 | read_length | total length of the read | int |
| 13 | mod_qual | probability of the base modification in the next column | int |
| 14 | mod_code | base modification code from the MM tag | str |
| 15 | base_qual | basecall quality score (phred) | int |
| 16 | ref_kmer | reference 5-mer sequence context (center base is aligned base), ‘.’ if unmapped | str |
| 17 | query_kmer | read 5-mer sequence context (center base is aligned base) | str |
| 18 | canonical_base | canonical base from the query sequence, from the MM tag | str |
| 19 | modified_primary_base | primary sequence base with the modification | str |
| 20 | inferred | whether the base modification call is implicit canonical | str |
| 21 | flag | FLAG from alignment record | str |
| 22 | motifs | comma-separated list of reference motifs matching at this position, only present when --motifs or --cpg is used | str |
Tabulating base modification calls for each read position with extract calls
The modkit extract calls command will generate a table of read-level base modification calls using the same thresholding algorithm employed by modkit pileup.
The resultant table has, for each read, one row for each base modification call in that read.
If a base is called as modified then call_code will be the code in the MM tag. If the base is called as canonical the call_code will be - (A, C, G, and T are
reserved for “any modification”). The full schema of the table is below:
| column | name | description | type |
|---|---|---|---|
| 1 | read_id | name of the read | str |
| 2 | forward_read_position | 0-based position on the forward-oriented read sequence | int |
| 3 | ref_position | aligned 0-based reference sequence position, -1 means unmapped | int |
| 4 | chrom | name of aligned contig, or ‘.’ if unmapped | str |
| 5 | mod_strand | strand of the molecule the base modification is on | str |
| 6 | ref_strand | strand of the reference the read is aligned to, or ‘.’ if unmapped | str |
| 7 | ref_mod_strand | strand of the reference with the base modification, or ‘.’ if unmapped | str |
| 8 | fw_soft_clipped_start | number of bases soft clipped from the start of the forward-oriented read | int |
| 9 | fw_soft_clipped_end | number of bases soft clipped from the end of the forward-oriented read | int |
| 10 | alignment_start | leftmost (i.e. smallest) aligned reference position | int |
| 11 | alignment_end | rightmost (i.e. largest) aligned reference position | int |
| 12 | read_length | total length of the read | int |
| 13 | call_prob | probability of the base modification call in the next column | int |
| 14 | call_code | base modification call, - indicates a canonical call | str |
| 15 | base_qual | basecall quality score (phred) | int |
| 16 | ref_kmer | reference 5-mer sequence context (center base is aligned base), ‘.’ if unmapped | str |
| 17 | query_kmer | read 5-mer sequence context (center base is aligned base) | str |
| 18 | canonical_base | canonical base from the query sequence, from the MM tag | str |
| 19 | modified_primary_base | primary sequence base with the modification | str |
| 20 | fail | true if the base modification call fell below the pass threshold | str |
| 21 | inferred | whether the base modification call is implicit canonical | str |
| 22 | within_alignment | when alignment information is present, is this base aligned to the reference | str |
| 23 | flag | FLAG from alignment record | str |
| 24 | motifs | comma-separated list of reference motifs matching at this position, only present when --motifs or --cpg is used | str |
Note on implicit base modification calls.
The . MM flag indicates that primary sequence bases without an associated base modification probability
should be inferred to be canonical. By default, when this flag is encountered in a modBAM, modkit extract will
output rows with the inferred column set to true and a mod_qual value of 0.0 for the base modifications
called on that read. For example, if you have a A+a. MM tag, and there are A bases in the read for which
there aren’t base modification calls (identifiable as non-0s in the MM tag) will be rows where the mod_code
is a and the mod_qual is 0.0.
Note on non-primary alignments
If a valid MN tag is found, secondary and supplementary alignments can be output in the modkit extract tables above.
See troubleshooting for details on how to get valid MN tags.
To have non-primary alignments appear in the output, the --allow-non-primary flag must be passed.
By default, the primary alignment will have all base modification information contained on the read, including soft-clipped and unaligned read positions.
If the --mapped-only flag is used, soft clipped sections of the read will not be included.
For secondary and supplementary alignments, soft-clipped positions are not repeated. See advanced usage for more details.
Tabulating per-read base modification content with extract read-stats.
Produce a table where modification counts are summarized on the read level.
This table will have one record (line) per valid read and count the number of modified and unmodified bases for each base modification specified on the command line.
To specify which modifications to use, pass --mod-codes {primary_base}:{mod_code} on the command line.
Multiple {primary_base}:{mod_code} pairs can be passed, separated by spaces.
For example --mod-codes C:m C:m will count the number of m (5mC) and h (5hmC) calls per-read.
Modification codes that are encountered, but not specified on the command line will be added to the other_modified_{primary_base} count.
For example, if you have direct RNA reads with m6A and Inosine calls you can use --mod-codes A:a and Modkit will count the m6A calls on each read and report it in the modified_a column.
Inosine calls will be counted in other_modified_A.
Using a filter-threshold
By default extract read-stats will simply tabulate the calls for each base modification on each read.
The “call” is defined as the state with the highest probability.
Passing --filter-threshold to extract read-stats will filter calls based on the probability, similar to what other functions do (see filtering modified-base calls for details).
A filter threshold for each primary sequence base can be used (e.g. --filter-threshold A:0.8 --filter-threshold C:0.9) which will use 0.8 and 0.9 for any base call on adenine or cytosine base, respectively.
To use a filter threshold for a specific base modification (e.g. for 6mA calls or 5hmC calls only) pass --mod-thresholds {mod_code}:{threshold} (e.g. --mod-thresholds a:0.9 or --mod-thresholds h:0.8).
When any threshold is used, extra columns are added to the output: fail_modified_{mod_code} and fail_unmodified_{primary_base}.
These columns contain the counts of modified or unmodified calls that are below the pass threshold for that base/modification.
Schema of output for modkit extract read-stats without filtering:
| column | name | description | type |
|---|---|---|---|
| 1 | read_id | name of the BAM record | str |
| 2 | chrom | name of the contig the read is mapped to or “unmapped” for unmapped reads | str |
| 3 | aln_start | leftmost aligned position of the read on the reference genome (-1 for unmapped reads) | int |
| 3+n_bases | unmodified_{A|C |G |T} | number of unmodified (canonical) calls for this primary sequebce base | int |
| 3+n_bases+n_mods | modified_{modification_code} | number of modified bases of this type on this read | int |
| 3+n_bases+n_mods+1+n_bases | other_modified_{A|C|G|T} | number of modified bases not specified on the command line | int |
| 3+2*n_bases+n_mods | read_length | length of SEQ in the BAM record | int |
Example usages:
Extract base modification counts for 5mC, 5hmC, and 6mA on each read in a BAM
modkit extract read-stats <input.bam> <output.csv> --mod-codes C:m C:h A:a
To enable filtering, use --filter-threshold (and optionally --mod-thresholds)
modkit extract read-stats <input.bam> <output.csv> --mod-codes C:m C:h A:a --filter-threshold 0.7 --mod-thresholds a:0.9
Extract base modification counts for m6A, Inosine, and pseudouridine from a direct RNA modBAM:
modkit extract read-stats <input.bam> <output.csv> --mod-codes A:a A:17596 T:17802
Extract a table of base modification probabilities from an aligned and indexed BAM
modkit extract full <input.bam> <output.tsv> [--bgzf]
If the index input.bam.bai can be found, intervals along the aligned genome can be performed
in parallel. The optional --bgzf flag will emit compressed output.
Extract a table from a region of a large modBAM
The below example will extract reads from only chr20, and include reference sequence context
modkit extract full <intput.bam> <output.tsv> --region chr20 --ref <ref.fasta>
Extract only sites aligned to a CG motif
modkit motif bed <reference.fasta> CG 0 > CG_motifs.bed
modkit extract full <in.bam> <out.tsv> --ref <ref.fasta> --include-bed CG_motifs.bed
Extract only sites that are at least 50 bases from the ends of the reads
modkit extract full <in.bam> <out.tsv> --edge-filter 50
Extract read-level base modification calls
modkit extract calls <input.bam> <calls.tsv>
Use --allow-non-primary to get secondary and supplementary mappings in the output.
modkit extract calls <input.bam> <output.tsv> --allow-non-primary
See the help string and/or advanced_usage for more details and performace considerations if you encounter issues with memory usage.
Investigating patterns with localise
One a bedMethyl table has been created, modkit localise will use the pileup and calculate per-base modification aggregate information around genomic features of interest.
For example, we can investigate base modification patterns around CTCF binding sites.
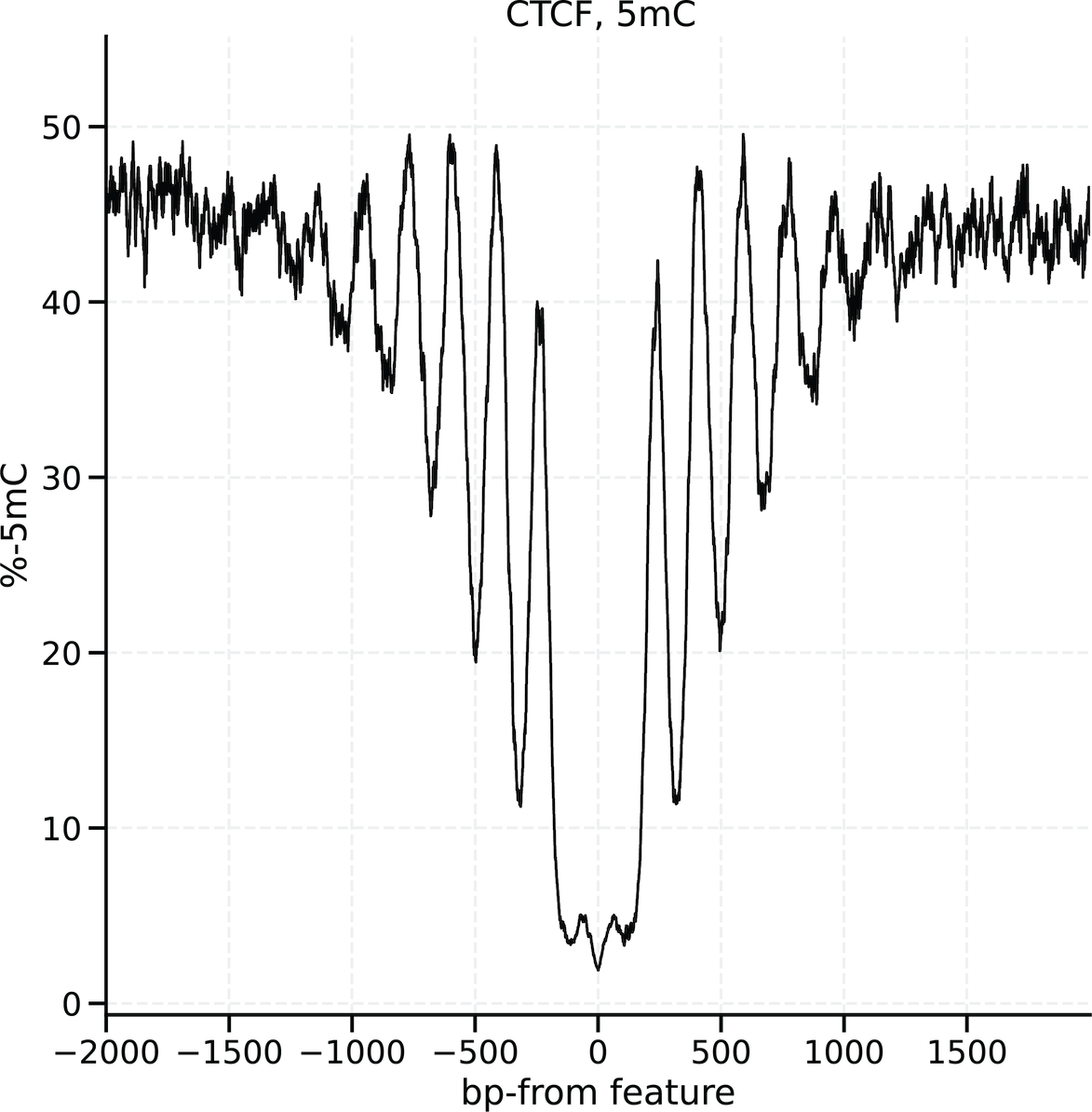
The input requirements to modkit localise are simple:
- BedMethyl table that has been bgzf-compressed and tabix-indexed
- Regions file in BED format (plaintext).
- Genome sizes tab-separated file:
<chrom>\t<size_in_bp>
an example command:
modkit localise ${bedmethyl} --regions ${ctcf} --genome-sizes ${sizes}
The output table has the following schema:
| column | Name | Description | type |
|---|---|---|---|
| 1 | mod code | modification code as present in the bedmethyl | str |
| 2 | offset | distance in base pairs from the center of the genome features, negative values reflect towards the 5’ of the genome | int |
| 3 | n_valid | number of valid calls at this offset for this modification code | int |
| 4 | n_mod | number of calls for this modification code at this offset | int |
| 5 | percent_modified | n_mod / n_valid * 100 | float |
Optionally the --chart argument can be used to create HTML charts of the modification patterns.
Perform differential methylation scoring
The modkit dmr command contains two subcommands, pair and multi, that will compare pairwise conditions and multiple conditions.
The pair command can be used to perform differential methylation detection on single genome positions (for example CpGs) or regions provided as a BED file.
On the other hand, multi can only be used to compare regions (such as CpG islands), provided as a BED file.
There are essentially three differential methylation workflows:
- Perform differential methylation scoring with a pair of samples on regions of the genome.
- Perform differential methylation scoring across all pairs of samples on regions of the genome.
- Perform base-level differential modification detection for a pair of conditions.
Each application is explained below. For details on the scoping of these applications see the limitations.
Preparing the input data
The inputs to all modkit dmr commands are two or more bedMethyl files (created by modkit pileup) that have been compressed with bgzip and indexed with tabix.
An example of how to generate the input data is shown below:
ref=grch38.fasta
threads=32
norm=normal_sample.bam
norm_pileup=normal_pileup.bed
modkit pileup ${norm} ${norm_pileup} \
--cpg \
--ref ${ref} \
--threads ${threads} \
--log-filepath log.txt
bgzip -k ${norm_pileup}
tabix -p bed ${norm_pileup}.gz
# pileup and compression can also be done in one step
tumor=tumor_sample.bam
tumor_pileup=tumor_pileup.bed.gz
modkit pileup ${tumor} - \
--cpg \
--ref ${ref} \
--threads ${threads} \
--log-filepath log.txt | ${bgzip} -c > ${tumor_pileup}
tabix -p bed ${tumor_pileup}
1. Perform differential methylation scoring of genomic regions for a pair of samples.
Once you have the two samples to be compared in the appropriate format, the final piece necessary is a BED file of the regions to be compared.
To continue with our example we can get CpG Islands from the UCSC table browser.
The data may not always be appropriate input for modkit.
For example, the CpG Islands track has extra columns and a header line:
#bin chrom chromStart chromEnd name length cpgNum gcNum perCpg perGc obsExp
660 chr20 9838623 9839213 CpG: 47 590 47 383 15.9 64.9 0.76
661 chr20 10034962 10035266 CpG: 35 304 35 228 23 75 0.85
Therefore, we need to transform the data with awk or similar, such as:
awk 'BEGIN{FS="\t"; OFS="\t"} NR>1 {print $2, $3, $4, $5}' cpg_islands_ucsc.bed \
| bedtools sort -i - > cpg_islands_ucsc_cleaned.bed
Keeping the name column is optional.
Sorting the regions isn’t strictly necessary, the output will be in the same order as the regions file.
Below is an example command to produce the scored output.
The --base option tells modkit dmr which bases to use for scoring the differences, the argument should be a canonical nucleotide (A, C, G, or T) whichever primary sequence base has the modifications you’re interested in capturing.
For example, for CpG islands the base we’re interested in is C.
regions=cpg_islands_ucsc_cleaned.bed
dmr_result=cpg_islands_tumor_normal.bed
modkit dmr pair \
-a ${norm_pileup}.gz \
--index-a ${norm_pileup}.gz.tbi \ # optional
-b ${tumor_pileup}.gz \
--index-b ${tumor_pileup}.gz.tbi \ # optional
-o ${dmr_result} \ # output to stdout if not present
-r ${regions} \
--ref ${ref} \
--base C \ # may be repeated if multiple modifications are being used
--threads ${threads} \
--log-filepath dmr.log
The full schema is described below with an example output.
2. Perform differential methylation detection on all pairs of samples over regions from the genome.
The modkit dmr multi command runs all pairwise comparisons for more than two samples for all regions provided in the regions BED file.
The preparation of the data is identical to that for the previous section (for each sample, of course).
Note that if multiple samples are given the same name, they will be combined.
An example command could be:
modkit dmr multi \
-s ${norm_pileup_1}.gz norm1 \
-s ${tumor_pileup_1}.gz tumor1 \
-s ${norm_pileup_2}.gz norm2 \
-s ${tumor_pileup_2}.gz tumor2 \
-o ${dmr_dir} \ # required for multi
-r ${cpg_islands} \ # skip this option to perform base-level DMR
--ref ${ref} \
--base C \
-t 10 \
-f \
--log-filepath dmr_multi.log
For example the samples could be haplotype-partitioned bedMethyl tables or biological replicates.
Unlike for modkit dmr pair a sample name (e.g. norm1 and tumor1 above) must be provided for each input
sample.
If you have many samples but a single reference sample you can use the --ref-sample option to specify the reference sample to be used for all comparisons. This is useful for large datasets with many samples where you are not interested in all pairwise comparisons to reduce computation.
modkit dmr multi \
-s ${norm_pileup_1}.gz norm1 \
-s ${tumor_pileup_1}.gz tumor1 \
-s ${tumor_pileup_2}.gz tumor2 \
-o ${dmr_dir} \ # required for multi
-r ${cpg_islands} \ # skip this option to perform base-level DMR
--ref ${ref} \
--ref-sample norm1 \ # use norm1 as the reference for all comparisons
--base C \
-t 10 \
-f \
--log-filepath dmr_multi.log
3. Detecting differential modification at single base positions
The modkit dmr pair command has the ability to score individual bases (e.g. differentially methylated CpGs).
To run single-base analysis on one or more paired samples, simply omit the --regions (-r) option when running modkit dmr pair.
When performing single-base analysis the likelihood ratio score and a MAP-based p-value are available.
For details on the likelihood ratio score and the MAP-based p-value, see the scoring details section.
For example the above command becomes:
dmr_result=single_base_haplotype_dmr.bed
modkit dmr pair \
-a ${hp1_pileup}.gz \
-b ${hp2_pileup}.gz \
-o ${dmr_result} \
--ref ${ref} \
--base C \
--threads ${threads} \
--log-filepath dmr.log
Multiple replicates can be provided as well by repeating the -a and -b options, such as:
dmr_result=tumor_normal_single_base_replicates.bed
modkit dmr pair \
-a ${norm_pileup_1}.gz \
-a ${norm_pileup_2}.gz \
-b ${tumor_pileup_1}.gz \
-b ${tumor_pileup_2}.gz \
-o ${dmr_result_replicates} \
--ref ${ref} \
--base C \
--threads ${threads} \
--log-filepath dmr.log
Keep in mind that the MAP-based p-value provided in single-site analysis is based on a “modified” vs “unmodified” model, see the scoring section and limitations for additional details.
Note about modification codes
The modkit dmr commands require the --base option to determine which genome positions to compare, i.e. --base C tells modkit to compare methylation at cytosine bases.
You may use this option multiple times to compare methylation at multiple primary sequence bases.
It is possible that, during pileup a read will have a mismatch and a modification call, such as a C->A mismatch and a 6mA call on that A, and you may not want to use that 6mA call when calculating the differential methylation metrics.
To filter out bedMethyl records like this, modkit uses the SAM specification (page 9) of modification codes to determine which modification codes apply to which primary sequence bases.
For example, h is 5hmC and applies to cytosine bases, a is 6mA and applies to adenine bases.
However, modkit pileup does not require that you use modification codes only in the specification.
If your bedMethyl has records with custom modification codes or codes that aren’t in the specification yet, use --assign-code <mod_code>:<primary_base> to indicate the code applies to a given primary sequence base.
Differential methylation output format
The output from modkit dmr pair (and for each pairwise comparison with modkit dmr multi) is (roughly) a BED file with the following schema:
| column | name | description | type |
|---|---|---|---|
| 1 | chrom | name of reference sequence from bedMethyl input samples | str |
| 2 | start position | 0-based start position, from --regions argument | int |
| 3 | end position | 0-based exclusive end position, from --regions argument | int |
| 4 | name | name column from --regions BED, or chr:start-stop if absent, “.” for single sites | str |
| 5 | score | difference score, more positive values have increased difference | float |
| 6 | strand | strand for the region or single-base position | str |
| 7 | samplea counts | counts of each base modification in the region, comma-separated, for sample A | str |
| 8 | samplea total | total number of base modification calls in the region, including unmodified, for sample A | int |
| 9 | sampleb counts | counts of each base modification in the region, comma-separated, for sample B | str |
| 10 | sampleb total | total number of base modification calls in the region, including unmodified, for sample B | int |
| 11 | samplea percents | percent of calls for each base modification in the region, comma-separated, for sample A | str |
| 12 | sampleb percents | percent of calls for each base modification in the region, comma-separated, for sample B | str |
| 13 | samplea fraction modified | fraction modification (of any kind) in sample A | float |
| 14 | sampleb fraction modified | fraction modification (of any kind) in sample B | float |
| (15) | cohen_h | Cohen’s h statistic (useful with regions and high-depth runs) | float |
| (16) | cohen_h_low | 95% confidence interval lower bound | float |
| (17) | cohen_h_high | 95% confidence interval upper bound | float |
an example of the output is given below:
#chrom start end name score strand a_counts a_total b_counts b_total a_mod_percentages b_mod_percentages a_pct_modified b_pct_modified effect_size cohen_h cohen_h_low cohen_h_high
chr20 9838623 9839213 CpG: 47 266.4940337502594 . h:12,m:45 1777 h:40,m:569 2101 h:0.68,m:2.53 h:1.90,m:27.08 0.032076534 0.28986198 -0.25778544 -0.7769052105982954 0.7137374830784644 0.8400729381181264
chr20 10034962 10035266 CpG: 35 5.451198465947527 . h:7,m:0 1513 h:8,m:8 1352 h:0.46,m:0.00 h:0.59,m:0.59 0.00462657 0.01183432 -0.00720775 -0.08185985199348272 0.008509433779133421 0.155210270207832
chr20 10172120 10172545 CpG: 35 7.8346692400409665 . h:15,m:28 1229 h:10,m:60 1091 h:1.22,m:2.28 h:0.92,m:5.50 0.034987796 0.06416132 -0.029173527 -0.1358646714824175 0.05433723875835786 0.21739210420647712
chr20 10217487 10218336 CpG: 59 185.80326974114905 . h:29,m:108 2339 h:38,m:453 1852 h:1.24,m:4.62 h:2.05,m:24.46 0.05857204 0.26511878 -0.20654674 -0.5928872447820701 0.5319235444885967 0.6538509450755435
chr20 10433628 10434345 CpG: 71 -0.06490108458092436 . h:29,m:2 2750 h:32,m:5 3737 h:1.05,m:0.07 h:0.86,m:0.13 0.0112727275 0.00990099 0.0013717376 0.013409890828678855 -0.03583284167510216 0.06265262333245987
chr20 10671925 10674963 CpG: 255 6.372129285562551 . h:43,m:24 9461 h:101,m:54 12880 h:0.45,m:0.25 h:0.78,m:0.42 0.0070817037 0.012034162 -0.004952458 -0.05133810350234183 0.024799813076449084 0.07787639392823457
n.b. Columns 15, 16, and 17 are present when the --regions option is passed, but these columns are on the right side of the table when performing single-site analysis (below).
It is generally recommended to use the --header flag and standard CSV parsing to make sure the schema’s between experiments are maintained.
When performing single-site analysis, the following additional columns are added:
| column | name | description | type |
|---|---|---|---|
| 15 | MAP-based p-value | ratio of the posterior probability of observing the effect size over zero effect size | float |
| 16 | effect size | percent modified in sample A (col 12) minus percent modified in sample B (col 13) | float |
| 17 | balanced MAP-based p-value | MAP-based p-value when all replicates are balanced | float |
| 18 | balanced effect size | effect size when all replicates are balanced | float |
| 19 | pct_a_samples | percent of ‘a’ samples used in statistical test | float |
| 20 | pct_b_samples | percent of ‘b’ samples used in statistical test | float |
| 21 | per-replicate p-values | MAP-based p-values for matched replicate pairs | float |
| 22 | per-replicate effect sizes | effect sizes matched replicate pairs | float |
| 23 | cohen_h | Cohen’s h statistic (useful with regions and high-depth runs) | float |
| 24 | cohen_h_low | 95% confidence interval lower bound | float |
| 25 | cohen_h_high | 95% confidence interval upper bound | float |
Columns 16-19 are only produced when multiple samples are provided, columns 20 and 21 are only produced when there is an equal number of ‘a’ and ‘b’ samples.
When using multiple samples, it is possible that not every sample will have a modification fraction at a position.
When this happens, the statistical test is still performed and the values of pct_a_samples and pct_b_samples reflect the percent of samples from each condition used in the test.
Columns 20 and 21 have the replicate pairwise MAP-based p-values and effect sizes which are calculated based on their order provided on the command line. For example in the abbreviated command below:
modkit dmr pair \
-a ${norm_pileup_1}.gz \
-a ${norm_pileup_2}.gz \
-b ${tumor_pileup_1}.gz \
-b ${tumor_pileup_2}.gz \
...
Column 20 will contain the MAP-based p-value comparing norm_pileup_1 versus tumor_pileup_1 and norm_pileup_2 versus norm_pileup_2.
Column 21 will contain the effect sizes, values are comma-separated.
If you have a different number of samples for each condition, such as:
modkit dmr pair \
-a ${norm_pileup_1}.gz \
-a ${norm_pileup_2}.gz \
-a ${norm_pileup_3}.gz \
-b ${tumor_pileup_1}.gz \
-b ${tumor_pileup_2}.gz \
these columns will not be present.
The full table when performing single-site analysis with equal numbers of samples in groups, when running modkit dmr pair, will have the following schema:
| column | name | description | type |
|---|---|---|---|
| 1 | chrom | name of reference sequence from bedMethyl input samples | str |
| 2 | start position | 0-based start position, from --regions argument | int |
| 3 | end position | 0-based exclusive end position, from --regions argument | int |
| 4 | name | name column from --regions BED, or chr:start-stop if absent, “.” for single sites | str |
| 5 | score | difference score, more positive values have increased difference | float |
| 6 | strand | strand for the region or single-base position | str |
| 7 | samplea counts | counts of each base modification in the region, comma-separated, for sample A | str |
| 8 | samplea total | total number of base modification calls in the region, including unmodified, for sample A | int |
| 9 | sampleb counts | counts of each base modification in the region, comma-separated, for sample B | str |
| 10 | sampleb total | total number of base modification calls in the region, including unmodified, for sample B | int |
| 11 | samplea percents | percent of calls for each base modification in the region, comma-separated, for sample A | str |
| 12 | sampleb percents | percent of calls for each base modification in the region, comma-separated, for sample B | str |
| 13 | samplea fraction modified | fraction modification (of any kind) in sample A | float |
| 14 | sampleb fraction modified | fraction modification (of any kind) in sample B | float |
| 15 | MAP-based p-value | ratio of the posterior probability of observing the effect size over zero effect size | float |
| 16 | effect size | percent modified in sample A (col 12) minus percent modified in sample B (col 13) | float |
| 17 | balanced MAP-based p-value | MAP-based p-value when all replicates are balanced | float |
| 18 | balanced effect size | effect size when all replicates are balanced | float |
Segmenting on differential methylation
When running modkit dmr without --regions (i.e. single-site analysis) you can generate regions of differential methylation on-the-fly using the segmenting hidden Markov model (HMM).
To run segmenting on the fly, add the --segment $segments_bed_fp option to the command such as:
dmr_result=single_base_haplotype_dmr.bed
dmr_segments=single_base_segements.bed
modkit dmr pair \
-a ${hp1_pileup}.gz \
-b ${hp2_pileup}.gz \
-o ${dmr_result} \
--segment ${dmr_segments} \ # indicates to run segmentation
--ref ${ref} \
--base C \
--threads ${threads} \
--log-filepath dmr.log
The default settings for the HMM are to run in “coarse-grained” mode which will more eagerly join neighboring sites, potentially at the cost of including sites that are not differentially modified within “Different” blocks.
To activate “fine-grained” mode, pass the --fine-grained flag.
The output schema for the segments is:
| column | name | description | type |
|---|---|---|---|
| 1 | chrom | name of reference sequence from bedMethyl input samples | str |
| 2 | start position | 0-based start position, from --regions argument | int |
| 3 | end position | 0-based exclusive end position, from --regions argument | int |
| 4 | state-name | “different” when sites are differentially modified, “same” otherwise | str |
| 5 | score | difference score, more positive values have increased difference | float |
| 6 | N-sites | number of sites (bedmethyl records) in the segment | float |
| 7 | samplea counts | counts of each base modification in the region, comma-separated, for sample A | str |
| 8 | sampleb counts | counts of each base modification in the region, comma-separated, for sample B | str |
| 9 | samplea percents | percent of calls for each base modification in the region, comma-separated, for sample A | str |
| 10 | sampleb percents | percent of calls for each base modification in the region, comma-separated, for sample B | str |
| 11 | samplea fraction modified | percent modification (of any kind) in sample A | float |
| 12 | sampleb fraction modified | percent modification (of any kind) in sample B | float |
| 13 | effect size | percent modified in sample A (col 11) minus percent modified in sample B (col 12) | float |
| 14 | cohen_h | Cohen’s h statistic (useful with regions and high-depth runs) | float |
| 15 | cohen_h_low | 95% confidence interval lower bound | float |
| 16 | cohen_h_high | 95% confidence interval upper bound | float |
Investigating differential modification on native RNA (direct RNA sequencing)
Modkit contains two functions for investigating methylation in direct RNA sequencing reads.
These two subcommands, dmr isoform and dmr compare-tx-sites, are meant to use a bedMethyl generated by pileup starting from a modBAM containing direct RNA reads aligned to a reference transcriptome, as opposed to a reference genome.
There are detailed instructions for how to run pileup in that command’s page.
Succinctly, a command similar to the one below will generate a bedMethyl table suitable for downstream use.
$ modkit pileup $modbam $output_pileup.bed.gz --modified-bases m6A --ref $transcriptome.fa --preload-references --bgzf
$ tabix $output_pileup.bed.gz
The following tools are available:
- Compare modifications between isoforms of genes
- Compare modifications between two conditions, aggregating isoform modifications on the gene level
Find positions varying in modification across isoforms with dmr isoform.
This command will compare the modification levels at common exonic positions for all isoforms of a gene.

Example
The input to dmr isoform is a single bedMethyl table that has been generated from a modBAM of reads aligned to a reference transcriptome (or transcriptome assembly).
This command also requires an accompanying GTF file with valid transcript models.
Below is an example command:
$ modkit dmr isoform \
${bedmethyl} \
isoform_dmr.bed \
--gtf ${gtf}
The bedMethyl table is expected to be bgzipped and have a tabix index.
To generate an isoform SVG plot like the one above, add the following arguments:
modkit dmr isoform \
${bedmethyl} \
isoform_dmr_${gene_name}.bed \
--plot path/to/plot/dir \
--gene-name ${gene_name} \
--gtf ${gtf}
Important:
--plotrequires either--gene-nameor--gene-idand cannot be used in whole-transcriptome mode (i.e. without one of those arguments). Attempting to use--plotwithout a gene specifier will result in an error.
Note that exons without marks in certain transcripts indicates that there is no data in the input bedMethyl.
The command modkit bedmethyl map-to-genome can map a transcript-aligned bedMethyl to genome coordinates.
Schema of output
| column | name | description | type |
|---|---|---|---|
| 1 | chrom | name of contig from GTF | str |
| 2 | chromStart | 0-based start position | int |
| 3 | chromEnd | 0-based exclusive end position | int |
| 4 | name | modification codes present at this position | str |
| 5 | score | likelihood ration score | float |
| 6 | strand | strand, ‘+’ or ‘-’ | str |
| 7 | p_value | p-value of alternative hypothesis | float |
| 8 | max_absolute_difference | largest effect size of all isoforms at this position | float |
| 9 | n_transcripts | number of transcripts (isoforms) contributing to this position | int |
| 10 | gene_id | gene-id from the GTF | str |
| 11 | gene_name | gene-name from the GTF or ‘-’ if not found | str |
| 12 | pooled_proportions | gene-level aggregate modification rate | str |
| 13 | per_isoform_proportions | JSON formatted string of per-transcript, per-modification proprotions | str |
| 14 | per_isoform_counts | JSON formatted string of per-transcript, per-modification counts | str |
Columns 12 and 14 are only present when the --full flag is passed.
Background
Gene sequences alone don’t describe all of the diversity of mRNAs in vertebrate cells. Through alternative splicing gene exons can be combined in multiple permutations called isoforms.

Filtering the number of methylation marks
Some long genes may have many modified positions and drawing them all can get crowded.
To only plot positions with a maximum p-value or minimum score use the --max-pval or
--min-score arguments, respectively.
Note:
--max-pvaland--min-scoreare plot-only options. They require--plot(and therefore also--gene-nameor--gene-id) and have no effect outside of single-gene mode. These two flags are mutually exclusive — only one may be provided at a time.
Compare methylation levels at transcriptome sites, summarized by gene, with dmr compare-tx-sites
The dmr compare-tx-sites allows you to compare modifications between two conditions.
This command uses a transcriptome-aligned bedMethyl table and maps the coordinates from each transcript to common gene coordinates.
After which, the modification counts from each isoform are combined making a gene-level pileup.
The gene-level modifications are then compared using the same method as dmr isoform.
Example command:
This command uses two bedMethyl tables corresponding to two conditions to be compared. These bedMethyl tables are expected to be bgzipped and have tabix indices.
modkit dmr compare-tx-sites \
-a ${bedmethyl_a} \
-b ${bedmethyl_b} \
--out ${output.bed} \
--gtf ${gtf}
A volcano plot can be also made, using the following extra options. An example is below.
modkit dmr compare-tx-sites \
-a ${bedmethyl_a} \
-b ${bedmethyl_b} \
--out ${output.bed} \
--single-code a \
--plot volcano.svg \
--gtf ${gtf} \
--log debug.log
The volcano plot requires that --single-mod-code is used, since log2 fold-change is essentially a 2-class metric.

These data were downloaded from the reference You et al., Benchmarking long-read RNA-sequencing technologies with LongBench: a cross-platform reference dataset profiling cancer cell lines with bulk and single-cell approaches
Schema of output
| column | name | description | type |
|---|---|---|---|
| 1 | chrom | name of contig from GTF | str |
| 2 | chromStart | 0-based start position | int |
| 3 | chromEnd | 0-based exclusive end position | int |
| 4 | name | modification codes present at this position | str |
| 5 | score | likelihood ration score | float |
| 6 | strand | strand, ‘+’ or ‘-’ | str |
| 7 | p_value | p-value of alternative hypothesis | float |
| 8 | gene_id | gene-id from the GTF | str |
| 9 | gene_name | gene-name from the GTF or ‘-’ if not found | str |
| 10 | log2_fold_change | log base 2 fold change | float |
| 11 | effect_size | difference in modification proportions between conditions | float |
| 12 | cond_a_proportions | JSON formatted string of condition ‘A’ per-modification proprotions | str |
| 13 | cond_b_proportions | JSON formatted string of condition ‘B’ per-modification proprotions | str |
| 14 | cond_a_counts | JSON formatted string of condition ‘A’ per-modification counts | str |
| 15 | cond_b_counts | JSON formatted string of condition ‘B’ per-modification counts | str |
Columns 11 through 15 are only present when the --full flag is passed.
Validating ground truth results.
The modkit validate sub-command is intended for validating results in a uniform manner from samples with known modified base content. Specifically the modified base status at any annotated reference location should be known.
Validating from modBAM reads and BED reference annotation.
The input to the modkit validate command will be pairs of modBAM and BED files.
modBAM files should contain modified base calls in the MM/ML tag as input to most modkit commands.
BED files paired to each input modBAM file describe the ground truth modified base status at reference positions.
This ground truth status should be known by the researcher due to previous experimental conditions and cannot be derived by modkit.
modkit validate \
--bam-and-bed sample1.bam sample1_annotation.bed \
--bam-and-bed sample2.bam sample2_annotation.bed
This will produce output such as the following:
> Parsing BED at /path/to/sample1_annotation.bed
> Processed 10 BED lines
> Parsing BED at /path/to/sample2_annotation.bed
> Processed 10 BED lines
> Canonical base: C
> Parsing mapping at /path/to/sample1.bam
> Processed 10 mapping recrods
> Parsing mapping at /path/to/sample2.bam
> Processed 10 mapping recrods
> Raw counts summary
Called Base
┌───┬───────┬───────┬───┬───┬───┬──────────┐
│ │ - │ a │ C │ G │ T │ Deletion │
├───┼───────┼───────┼───┼───┼───┼──────────┤
Ground │ - │ 9,900 │ 100 │ 1 │ 1 │ 1 │ 2 │
Truth │ a │ 100 │ 9,900 │ 1 │ 1 │ 1 │ 2 │
└───┴───────┴───────┴───┴───┴───┴──────────┘
> Balancing ground truth call totals
> Raw accuracy: 99.00%
> Raw modified base calls contingency table
Called Base
┌───┬────────┬────────┐
│ │ - │ a │
├───┼────────┼────────┤
Ground │ - │ 99.00% │ 1.00% │
Truth │ a │ 1.00% │ 99.00% │
└───┴────────┴────────┘
> Call probability threshold: 0.95
> Filtered accuracy: 99.90%
> Filtered modified base calls contingency table
Called Base
┌───┬────────┬────────┐
│ │ - │ a │
├───┼────────┼────────┤
Ground │ - │ 99.90% │ 0.10% │
Truth │ a │ 0.10% │ 99.90% │
└───┴────────┴────────┘
The filtering threshold is computed in the same manner as in all other modkit commands.
Currently only a defined percentage of input data filtering threshold estimation is implemented.
The default value is 10% (as in other modkit commands) and can be adjusted with the --filter-quantile argument.
Other methods (including user-defined thresholds) will be implemented in a future version.
The Call probability threshold is intended a value to be used for user-defined thresholds for other modkit commands.
BED ground truth annotation file:
A BED file is a tab-delimited file. For this command the first 6 fields are processed. These fields are as follows:
| column | name | description | |
|---|---|---|---|
| 1 | chrom | name of reference sequence | str |
| 2 | start position | 0-based start position | int |
| 3 | end position | 0-based exclusive end position | int |
| 4 | mod code | modified base code | str |
| 6 | strand | strand (e.g. +,-,.) | str |
The 5th column is ignored in the validate command.
The 4th column represents the modified base code annotating the status at this reference position (or range of reference positions).
This value can be - representing a canonical base (note that this differs from the remora validate annotation), a single letter code as defined in the modBAM tag specification, or any ChEBI code.
The validate command will assume that any base from the associated modBAM file overlapping these positions should match this annotation.
Output file
The --out-filepath option is provided to allow persistent storage of results in a machine-parseable format without other logging lines.
This format outputs all contingency tables in a machine-parseable format.
For example this contingency table [["ground_truth_label","-","a"],["-",9900,100],["a",100,9900]] would be produced from the above example results.
Calculating methylation entropy
The modkit entropy command will calculate the methylation entropy in genomic windows of defined length across the genome, optionally summarizing these calculations for regions.
Methylation entropy (ME) is a measure of the “information content” in the patterns of methylation reported by the sequencing reads, it could also be thought of as a measure of the randomness in the epialleles (an “epiallele” is the DNA modification at a given position).
This metric was originally proposed by Xie et al. and has been shown to correlation with regulation, aging, and cancer.
Unlike the pileup method in modkit which aggregates the modification calls per genomic position, modkit entropy looks at the co-occurrence of methylation status on individual reads and so the input is a modBAM not a pileup.
To quote Lee et al., “This information is important because such ‘phased’ methylation states can inform us about the epigenetic diversity of cell populations as well as the local regulation states of the epigenome”.
Probably the simplest visual description of methylation entropy is the following, a version of which appears in many of the methods papers:
Citation: Lee et al.
{kind=link}
Calculate entropy in windows across the genome
modkit entropy --in-bam ${mod_bam} \
-o ${output_entropy_bedgraph} \
--ref ${ref} \
--cpg \
--threads 32 \
--log-filepath modkit_entropy.log
When the output file, -o, is omitted the output will be to stdout.
Output schema
| column | name | description | type |
|---|---|---|---|
| 1 | chrom | contig name | string |
| 2 | start | start of interval | int |
| 3 | end | end of interval | int |
| 4 | entropy | methylation entropy | float |
| 5 | num_reads | number of reads used | int |
Calculating entropy in BED-specified regions
The command can also summarize the methylation entropy in regions by using the --regions option, for example:
modkit entropy \
--in-bam ${mod_bam} \
-o ${output_directory} \
--regions ${regions_bed_file} \ # BED3 or BED4 file of regions
--cpg \ # specify CpG dinucleotides and combine strands
--ref ${ref} \
--threads 32 \
--log-filepath modkit_entropy.log
The output must now be a directory (specified with -o), a bedGraph with the entropy over the windows with the regions as well as a summary of the methylation entropy in the regions will be output.
By default these files will be regions.bed and windows.bedgraph.
The schema for the regions.bed file is below:
| col | Name | Description | type |
|---|---|---|---|
| 1 | chrom | chromosome of the region | str |
| 2 | start | 0-based start position of the region | int |
| 3 | end | 0-based end position of the region | int |
| 4 | region_name | name of the region from the input BED file | str |
| 5 | mean_entropy | average entropy of the passing windows included in the region | float |
| 6 | strand | strand of the region {+, -, . } | str |
| 7 | median_entropy | median entropy of the passing windows included in the region | float |
| 8 | min_entropy | minimum passing window entropy | float |
| 9 | max_entropy | maximum passing window entropy | float |
| 10 | mean_num_reads | average number of reads used in the passing windows’ entropy calculation | float |
| 11 | min_num_reads | minimum number of reads used in the passing windows’ entropy calculation | int |
| 12 | max_num_reads | minimum number of reads used in the passing windows’ entropy calculation | int |
| 13 | successful_window_count | number of passing windows in the region | int |
| 14 | failed_window_count | number of failed windows in the region | int |
Specifying motifs or primary sequence bases
Similar to pileup you can specify a motif on the command line with --motif and optionally combine the counts across the positive and negative strands with --combine-strands.
If you specify a primary sequence base (with --base) or a motif (with --motif) that is not reverse-complement palindromic modkit will output methylation entropy per-strand.
This option can be passed multiple times to use multiple motifs.
When motifs specify different primary sequence bases, for example --motif GATC 1 and --motif CCGG 0, all of the base modification statuses will be used in calculating the entropy values.
For example if you want to calculate m6A entropy in DRACH motifs:
${modkit} entropy ${bam} \
-o ${output} \
--regions ${regions} \
--ref ${ref} \
--motif DRACH 2 \
--threads 32 \
--log-filepath modkit_entropy.log \
When performing transcriptome analysis, it’s recommended to make a regions BED file of all of the transcripts so that you can rank which transcripts have highest entropy.
Calculation of methylation entropy
The calculation of methylation entropy has been described in the papers linked above. Formally, methylation entropy in modkit is calculated as:
\[ \text{ME} = \frac{-1}{N} \sum_{\textbf{N}} Pr(n_i) * \text{log}_{2}Pr(n_i) \]
Where \( \textbf{N} \) is the set of all methylation patterns and \( Pr(n_i) \) is the empirical probability of that pattern.
To account for the fact that modkit filters base modification calls when they are below a certain confidence level, filtered positions are given a “wildcard” assignment and can match any epiallele at that position.
The entropy calculation implementation in modkit will assign a fractional count to each pattern that the read matches.
For example, suppose a read with epiallele m*mm meaning there are 4 positions in the window (5mC) and this read reports 5mC, followed by a filtered call, and 2 more 5mC calls.
This read will match to patterns [mhmm mmmm mCmm] (m = 5mC, h = 5hmC, and C is canonical cytosine).
When the --num-positions parameter gets large the number of potential patterns becomes large.
Most patterns will probably not have any reads matching to them, so instead of enumerating all possible patterns modkit uses a prefix trie to find all patterns represented in the reads while accounting for filtered positions.
Narrow output to specific positions
The pileup, sample-probs, summary, and extract sub commands have a --include-bed (or --include-positions) option that will restrict analysis to only positions that overlap with the intervals contained within the BED file.
In the case of pileup, summary, and sample-probs, the pass-threshold will be estimated with only base modification probabilities that are aligned to positions overlapping intervals in the BED. In the case of pileup and extract only positions will be reported if they overlap intervals in the BED.
Merge multiple bedMethyl files
If you generate bedMethyl tables from multiple samples and want to perform a “join” where the counts summed modkit bedmethyl merge will perform an outer join for you.
An example command is below:
modkit bedmethyl merge sample_a.bed.gz sample_b.bed.gz \
-o samples_a_and_b.bed \
-g genome_sizes.tsv # could be reference.fa.fai
The input bedMethyl files must be bgzipped and tabix indexed.
bgzip sample_a.bed
tabix sample_a.bed.gz
bgzip sample_b.bed
tabix sample_b.bed.gz
The genome_sizes.tsv is a tab-separated file containing the lengths of the contigs in the reference sequence, you can also use a .fai file generated by samtools faidx.
For example taken from bedtools:
The schema should be <chromName><TAB><chromSize>.
For example, Human (hg19):
chr1 249250621
chr2 243199373
Requiring positions in multiple samples (inner join)
By default merge performs an outer join: a position is kept if it appears in
any input. To instead keep only positions shared across inputs, use
--min-samples, which outputs a position only when it appears in at least that
many input files. Pass --min-samples all to require the position in every
input (an inner join), which is useful for retaining positions that reproduce
across replicates; all avoids hard-coding the input count in scripts.
# keep only positions present in all three replicates (inner join)
modkit bedmethyl merge rep1.bed.gz rep2.bed.gz rep3.bed.gz \
-o replicates_inner.bed \
-g genome_sizes.tsv \
--min-samples all
--min-sample-coverage adds a per-input confidence floor: an input only counts
towards a position (both for the --min-samples tally and for the summed counts)
when that input’s record has at least the given valid coverage. For example, to
require a position to be covered by at least 5 valid reads in all three
replicates:
modkit bedmethyl merge rep1.bed.gz rep2.bed.gz rep3.bed.gz \
-o replicates_inner_cov5.bed \
-g genome_sizes.tsv \
--min-samples all \
--min-sample-coverage 5
Omitting both options performs an outer-join.
Convert bedMethyl to bigWig
You may want to convert a bedMethyl to bigWig for easier visualization on a genome browser.
Modkit provides a bedmethyl tobigwig function that can extract a bigWig track from a bedMethyl.
The bigWig format is more constrained than bedMethyl in that it can only display a single stream of data (floats).
So you can’t have a bigWig with 5hmC and 5mC values together, you need to make separate tracks. Here are some annotated examples:
# you can run pileup and stream the output through the new command '-' means output to stdout
modkit pileup ${modbam} - \
# --mod-code a will make a track of 6mA
| tee >(modkit bedmethyl tobigwig - ${outdir}/subsample_pileup_a.bw --mod-code a -g ${sizes} --log ${outdir}/bm_a.log --negative-strand-values --suppress-progress) \
# you can use "bm" or "bedmethyl"
# --mod-code (or --mod-codes h,m) will combine the 5hmC and 5mC counts into a track
| tee >(modkit bm tobigwig - ${outdir}/subsample_pileup_hm.bw --mod-codes h,m -g ${sizes} --log ${outdir}/bm_hm.log --negative-strand-values --suppress-progress) \
# finally pipe out to the pileup
> ${pileup}
# you can use a file also, if you have conflicting bases (e.g. 6mA and 5mC) - you'll get an error.
modkit bm tobigwig ${pileup} ${outbw} --mod-code m,a -g ${sizes} --negative-strand-values
With this CLI you can capture any number of bigWig tracks whilst making a bedMethyl pileup at the same time. You can also make a bigWig from a pre-computed bedMethyl (and use tabix to get just sections of a pre-made bedMethyl).
ModBam Utilities
Checking MM/ML tags
In general, when Modkit encounters an record that it cannot use due to the modified base information being invalid or not conforming to the specification it will log the error and ignore the record.
However, it may be important to know precisely how many records are unusable and for what reason.
The modkit modbam check-tags command is for this purpose.
It will tabulate the error conditions in the modBAM and report out how many of each problem it encounters.
When there are any records that cause an error, the command will exit 1 (unless the --permissive flag is given).
You can use this command as part of a workflow to exit early if a modBAM is invalid.
Example usage
modkit modbam check-tags ${modbam} -t 20 --log ${outdir}/check_tags.log
# alternatively use the abbreviated subcommand
modkit mb check-tags ${modbam} -t 20 --log ${outdir}/check_tags.log
The command outputs the following tables
- Error counts, each error encountered and the number of records with the error. The
pctcolumn shows the percent of errors.
+-----------------------------------------+-------+-------+
| error | count | pct |
+-----------------------------------------+-------+-------+
| conflict-explicit-and-inferred | 3792 | 92.71 |
| conflict-explicit-prob-greater-than-one | 298 | 7.29 |
| total | 4090 | 100 |
+-----------------------------------------+-------+-------+
- Tag-header tables, counts of how many of each MM-tag “header” are found, e.g. “C+m.” means cytosine is the fundamental base, on the positive strand, and the “mode” is ‘.’ meaning that cytosines without explicit probabilities are inferred to be un-modified. See the specification for more details on the “mode”. There is one table for each the valid and invalid records.
+------------+-------+
| tag_header | count |
+------------+-------+
| C+m. | 4090 |
| A+a. | 4090 |
| N+e? | 4090 |
| C+h. | 4090 |
+------------+-------+
- Modified bases, a table of which primary sequence bases have which modification codes assigned to them as well as the “mode” in the tag corresponding to that assignment.
+--------+--------------+----------+------+
| strand | primary_base | mod_code | mode |
+--------+--------------+----------+------+
| + | T | b | ? |
| + | T | e | ? |
| + | G | b | ? |
| + | G | e | ? |
| + | C | b | . |
| + | C | e | . |
| + | C | h | . |
| + | C | m | . |
| + | A | a | . |
| + | A | b | . |
| + | A | e | . |
+--------+--------------+----------+------+
Find regions of accessible chromatin
Nanopore sequencing can detect multiple base modifications simultaneously and we can leverage this capability by introducing exogenous base modifications at specific functional regions. One such method uses a 6mA methyltransferase such as EcoGII or Hia5 to label accessible regions of chromatinized DNA, usually by treatment of cell nuclei with the enzyme.

Predict regions of open chromatin
Modkit comes with a machine learning model that has been trained to identify regions of open chromatin based on 6mA signal. You can invoke this model with the following command:
$ modkit open-chromatin predict ${mod_bam} \
--model ${model} \
--log modkit_predict.log \
-o ./accessible_regions.bedgraph \
--device 0
Where ${model} is the path to the directory with the model, for example dist_modkit_v0.5.0_38fda16/models/r1041_e82_400bps_hac_v5.2.0@v0.1.0.
The output of the command is a bedGraph file with the following schema:
| column | name | description | type |
|---|---|---|---|
| 1 | chrom | contig or scaffold | string |
| 2 | start | start of the region | int |
| 3 | end | end of the region | int |
| 4 | probability | accessibility probability, (0.0, 1) | float |
Configuring --step-size
The open chromatin model in Modkit works by making predictions on 100 base pair windows of the genome.
The output of the model is a prediction probability between 0.0 and 1.0, non-inclusive.
To make predictions on the whole genome (or regions when the --include-bed option is provided) Modkit applies the model to overlapping windows.
The --step-size determines how much to advance before making another prediction:
Genome <--//------------------//->
Window 1 |---w----|
Window 2 |_s_|---w----|
In the above w is the window size (100bp with the current model) and s is the step size which can be configured via the --step-size parameter.
The smaller the step size, the finer-graned resolution the output at the cost of more computation.
This can be seen in the following example browser image:

Step size of 25 base pairs is the default.
Using --threshold to get regions of high confidence
As seen above, the output from ope-chromatin predict is a stream of probabilities over the genome or desired region of the genome.
A lot of these predictions are going to be very close to zero.
You can remove low-probability regions with the --threshold option so that only intervals with a probability greater than or equal to this value are reported.
Using this option in combination with bedtools merge can transform the bedGraph file into a BED of predicted open chromatin regions.
$ modkit open-chromatin predict ${bam} \
--model ${model} \
--threshold 0.8 \
--include-bed promoters_slop2000.bed \
-o stdout \
| bedtools merge -i - > accessible_regions.bed
Running with a GPU
Modkit comes distributed with the capability to run open chromatin prediction on a normal CPU, NVIDIA GPUs, and can be built to run on Apple GPUs.
The normal distribution only has the capability to run on CPU hardware.
At the current stage of development this configuration is probably too slow for most practical purposes outside of small region checks.
The candle distribution can be downloaded and used with NVIDIA GPUs directly, however we cannot guarantee that it will work on every GPU setup.
For the best performance on GPU or CPU, the tch (pytorch) distribution is recommended, however it requires that you download libtorch from the internet.
Instructions can be found in the BUILD_NOTES_*.txt packaged with the software.
Troubleshooting: Checking your input data
To predict regions of open chromatin, the input modBAM should have 6mA base modification calls.
You can quickly check that you have valid 6mA calls using the check-tags command:
$ modkit modbam check-tags ${bam} --head 1000
This command will return non-zero when you have invalid MM/ML SAMTags. You should expect to see an output similar to this:
> checking tags on first 1000 reads
> no errors
> num PASS records: 1000 (100.00%)
> num records: 1000
> valid record tag headers:
+------------+-------+
| tag_header | count |
+------------+-------+
| A+a. | 1000 |
+------------+-------+
> modified bases:
+--------+--------------+----------+------+
| strand | primary_base | mod_code | mode |
+--------+--------------+----------+------+
| + | A | a | . |
+--------+--------------+----------+------+
modkit, subcommand documentation
The goal of modkit is to enable best-practices manipulation of BAM files containing
modified base information (modBAMs). The various sub-commands and tools available in
modkit are described below. This information can be obtained by invoking the long help
(--help) for each command.
Advanced usage information.
Modkit is a bioinformatics tool for working with modified bases from Oxford
Nanopore
Usage: modkit <COMMAND>
Commands:
pileup Tabulates base modification calls across genomic positions.
This command produces a bedMethyl formatted file. Schema and
description of fields can be found in the README
extract Extract read-level base modification information from a modBAM
into a tab-separated values table
dmr Perform DMR test on a set of regions. Output a BED file of
regions with the score column indicating the magnitude of the
difference. Find the schema and description of fields can in
the README as well as a description of the model and method.
See subcommand help for additional details
pileup-hemi Tabulates double-stranded base modification patters (such as
hemi-methylation) across genomic motif positions. This command
produces a bedMethyl file, the schema can be found in the
online documentation
validate Validate results from a set of mod-BAM files and associated
BED files containing the ground truth modified base status at
reference positions
motif Various commands to search for, evaluate, or further regine
sequence motifs enriched for base modification. Also can
generate BED files of motif locations
entropy Use a mod-BAM to calculate methylation entropy over genomic
windows
localize Investigate patterns of base modifications, by aggregating
pileup counts "localized" around genomic features of interest
stats Calculate base modification levels over regions
bedmethyl Utilities to work with bedMethyl files
modbam Utilities to work with modBAM files
open-chromatin Identify regions of open chromatin based on exogenous 6mA
signal
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help
-V, --version Print version
pileup
Tabulates base modification calls across genomic positions. This command
produces a bedMethyl formatted file. Schema and description of fields can be
found in the README
Usage: modkit pileup [OPTIONS] <IN_BAM> <OUT_BED>
Arguments:
<IN_BAM>
Input BAM, should be sorted and have associated index available
<OUT_BED>
Output file to write results into. Specify "-" or "stdout" to direct
output to stdout
Options:
--duplex
Specify that modBAM contains duplex modification calls and these
should be used
--modified-bases <MODIFIED_BASES>...
Specify which modified bases to tablulate counts for. These can be the
"long name" such as '5mC', '6mA' (or 'm6A' for RNA), or 'inosine'. You
can also pass <primary_base>:<mod_code>, such as 'C:m'. Finally, when
running with --combine-mods the reference base can be passed alone,
such as 'C' or 'A'
-h, --help
Print help (see a summary with '-h')
Output Options:
--bgzf
Output bgzf-compressed bedmethyl files suitable for Tabix-indexing
--header
Output bedMethyl where the delimiter of columns past column 10 are
space-delimited instead of tab-delimited. This option can be useful
for some browsers and parsers that don't expect the extra columns of
the bedMethyl format. Output a header with the bedMethyl
--prefix <PREFIX>
Prefix to prepend on phased output file names. Without this option the
files will be <mod_code>_<strand>.bedmethyl
--phased
Produce separate bedMethyl tables for haplotype-labeled modBAM
records. Currently only supports diploid genomes. Produces
hp1.bedmethyl, hp2.bedmethyl, and combined.bedmethyl files (optionally
compressed). hp1.bedmethyl and hp2.bedmethyl contain counts for
records with HP=1 and HP=2 tags, respectively. combined.bedmethyl
contains counts for all modBAM records
--bedrmod
Output BedRModV2 header counts based on V2 Specification. Details can
be found at https://dieterich-lab.github.io/euf-specs/bedRModv2.pdf
Compute Options:
--bgzf-threads <BGZF_THREADS>
Specify the number of threads to dedicate to BGZF compression
[default: 4]
-t, --threads <THREADS>
Number of threads to use while processing chunks concurrently
[default: 4]
--io-threads <IO_THREADS>
Number of IO/decompression threads to use
[default: 4]
-s, --sampling-threads <SAMPLING_THREADS>
Use this many threads when performing threshold estimation, setting
this to a higher value can speed up execution
-i, --interval-size <INTERVAL_SIZE>
Interval chunk size in base pairs to process concurrently. Smaller
interval chunk sizes will use less memory but incur more overhead
[default: 1000000]
--queue-size <QUEUE_SIZE>
Size of queue for writing records, default will be the number of
threads
--max-depth <MAX_DEPTH>
Maximum number of reads to use whilest tabulating counts at a given
position
[default: 8000]
--high-depth
Flag to indicate that modBAM has exceptionally high depth (>65,000X)
and should be downsampled during pileup
Logging Options:
--log-filepath <LOG_FILEPATH>
Specify a file for debug logs to be written to, otherwise ignore them.
Setting a file is recommended. (alias: log)
--suppress-progress
Hide the progress bar
Selection Options:
--region <REGION>
Process only the specified region of the BAM when performing pileup.
Format should be <chrom_name>:<start>-<end> or <chrom_name>. Commas
are allowed
--include-bed <INCLUDE_BED>
BED file that will restrict threshold estimation and pileup results to
positions overlapping intervals in the file. (alias:
include-positions)
--include-unmapped
Include unmapped base modifications when estimating the pass threshold
--edge-filter <EDGE_FILTER>
Discard base modification calls that are this many bases from the
start or the end of the read. Two comma-separated values may be
provided to asymmetrically filter out base modification calls from the
start and end of the reads. For example, 4,8 will filter out base
modification calls in the first 4 and last 8 bases of the read
Sampling Options:
-n, --num-reads <NUM_READS>
Sample this many reads when estimating the filtering threshold. Reads
will be sampled evenly across aligned genome. If a region is
specified, either with the --region option or the --sample-region
option, then reads will be sampled evenly across the region given.
This option is useful for large BAM files. In practice, 10-50 thousand
reads is sufficient to estimate the model output distribution and
determine the filtering threshold
[default: 10042]
--strict-sampling
Fail if we cannot sample at least the requested number of reads when
estimating the pass threshold
-f, --sampling-frac <SAMPLING_FRAC>
Sample this fraction of the reads when estimating the pass-threshold.
In practice, 10-100 thousand reads is sufficient to estimate the model
output distribution and determine the filtering threshold. See
filtering.md for details on filtering
--seed <SEED>
Set a random seed for deterministic running, the default is
non-deterministic
Filtering Options:
--no-filtering
Do not perform any filtering, include all mod base calls in output.
See filtering.md for details on filtering
-p, --filter-percentile <FILTER_PERCENTILE>
Filter out modified base calls where the probability of the predicted
variant is below this confidence percentile. For example, 0.1 will
filter out the 10% lowest confidence modification calls
[default: 0.1]
--filter-threshold <FILTER_THRESHOLD>
Specify the filter threshold globally or per-base. Global filter
threshold can be specified with by a decimal number (e.g. 0.75).
Per-base thresholds can be specified by colon-separated values, for
example C:0.75 specifies a threshold value of 0.75 for cytosine
modification calls. Additional per-base thresholds can be specified by
repeating the option: for example --filter-threshold C:0.75
--filter-threshold A:0.70 or specify a single base option and a
default for all other bases with: --filter-threshold A:0.70
--filter-threshold 0.9 will specify a threshold value of 0.70 for
adenine and 0.9 for all other base modification calls
--mod-threshold <MOD_THRESHOLDS>
Specify a passing threshold to use for a base modification,
independent of the threshold for the primary sequence base or the
default. For example, to set the pass threshold for 5hmC to 0.8 use
`--mod-threshold h:0.8`. The pass threshold will still be estimated as
usual and used for canonical cytosine and other modifications unless
the `--filter-threshold` option is also passed. See the online
documentation for more details
--max-filter-threshold <MAX_FILTER_THRESHOLD>
Maximum threhsold value to use, if the estimated threshold value for
any primary sequence base is greater than this value, use this value
instead. To set a default value for all primary bases, use
--max-threshold 0.8, for example. Or use per-base threshold values
like --max-threshold C:0.8
--sample-region <SAMPLE_REGION>
Specify a region for sampling reads from when estimating the threshold
probability. If this option is not provided, but --region is provided,
the genomic interval passed to --region will be used. Format should be
<chrom_name>:<start>-<end> or <chrom_name>
--sampling-interval-size <SAMPLING_INTERVAL_SIZE>
Interval chunk size in base pairs to process concurrently when
estimating the threshold probability, can be larger than the pileup
processing interval
[default: 1000000]
Modified Base Options:
--motif <MOTIF> <MOTIF>
Output pileup counts for only sequence motifs provided. The first
argument should be the sequence motif and the second argument is the
0-based offset to the base to pileup base modification counts for. For
example: --motif CGCG 0 indicates to pileup counts for the first C on
the top strand and the last C (complement to G) on the bottom strand.
The --cpg argument is short hand for --motif CG 0.
This argument can be passed multiple times. When more than one motif
is used, the resulting output BED file will indicate the motif in the
"name" field as <mod_code>,<motif>,<offset>. For example, given
`--motif CGCG 2 --motif CG 0` there will be output lines with name
fields such as "m,CG,0" and "m,CGCG,2". To use this option with
`--combine-strands`, all motifs must be reverse-complement palindromic
or an error will be raised.
--cpg
Only output counts at CpG motifs
-r, --reference <REFERENCE_FASTA>
Reference sequence in FASTA format. Required for motif (e.g. CpG)
filtering, requires FAI fasta index to be pre-generated. (alias:
'ref')
--preload-references
Preload the reference sequences, useful when working with many, short
reference sequences such as a transcriptome
-k, --mask
Respect soft masking in the reference FASTA
--combine-mods
Combine base modification calls, all counts of modified bases are
summed together. See collapse.md for details
--allow-non-primary
Use non-primary alignments in pileup (e.g. secondary and
supplementaty). Keep in mind these mappings will be used _in addition_
to the primary mappings
--combine-strands
When performing motif analysis (such as CpG), sum the counts from the
positive and negative strands into the counts for the positive strand
position. Requires that the BAM records have correct MN tags
BedRMod Options:
--organism <ORGANISM>
NCBI Taxonomic identifier, details at:
https://doi.org/10.1093/database/baaa062
[default: unknown]
--modification-type <MODIFICATION_TYPE>
A valid RNA type
[default: RNA]
--assembly <ASSEMBLY>
Genome assembly
[default: unknown]
--annotation-source <ANNOTATION_SOURCE>
Annotation source
[default: unknown]
--annotation-version <ANNOTATION_VERSION>
Annotation version
[default: unknown]
--sequencing-platform <SEQUENCING_PLATFORM>
Sequencing platform
[default: ont]
--basecalling <BASECALLING>
Basecalling model, override model set in BAM header
--bioinformatics-workflow <BIOINFORMATICS_WORKFLOW>
Link to bioinformatics workflow; program name, version, and/or call;
information relevant to score, coverage, or frequency calculation; etc
--experiment <EXPERIMENT>
Information about or link to experimental protocol and design
--external-source <EXTERNAL_SOURCE>
Databank:ID of data
--modomics-code <MODOMICS_CODE> <MODOMICS_CODE> <MODOMICS_CODE>
User-specified code, format: <Code> <MODOMICS Short Name> <Primary
Base>. <Code> is the base modification code described in the SAMtags
or a numeric ChEBI code present in the BAM file. For example 'a m6A A'
would be for <Code> 'a', <MODOMICS Short Name> m6A, and <Primary Base>
A
adjust-mods
Performs various operations on BAM files containing base modification
information, such as converting base modification codes and ignoring
modification calls. Produces a BAM output file
Usage: modkit adjust-mods [OPTIONS] <IN_BAM> <OUT_BAM>
Arguments:
<IN_BAM>
Input BAM file, can be a path to a file or one of `-` or `stdin` to
specify a stream from standard input
<OUT_BAM>
File path to new BAM file to be created. Can be a path to a file or
one of `-` or `stdin` to specify a stream from standard output
Options:
-f, --ff
Fast fail, stop processing at the first invalid sequence record.
Default behavior is to continue and report failed/skipped records at
the end
-h, --help
Print help (see a summary with '-h')
Output Options:
--log-filepath <LOG_FILEPATH>
Output debug logs to file at this path
--output-sam
Output SAM format instead of BAM
Modified Base Options:
--ignore <IGNORE>
Modified base code to ignore/remove, see
https://samtools.github.io/hts-specs/SAMtags.pdf for details on the
modified base codes
--convert <CONVERT> <CONVERT>
Convert one mod-tag to another, summing the probabilities together if
the retained mod tag is already present
--motif <MOTIF> <MOTIF>
Filter out any base modification call that isn't part of a basecall
sequence motif. This argument can be passed multiple times. Format is
<motif_sequence> <offset>. For example the argument to match CpG
dinucleotides is `--motif CG 0`, or to match CG[5mC]G the argument
would be `--motif CGCG 2`. Single bases can be used as motifs to keep
only base modification calls for a specific primary base, for example
`--motif C 0`
--cpg
Shorthand for --motif CG 0
--discard-motifs
Discard base modification calls that match the provided motifs
(instead of keeping them)
Compute Options:
-t, --threads <THREADS>
Number of threads to use
[default: 4]
Selection Options:
--edge-filter <EDGE_FILTER>
Discard base modification calls that are this many bases from the
start or the end of the read. Two comma-separated values may be
provided to asymmetrically filter out base modification calls from the
start and end of the reads. For example, 4,8 will filter out base
modification calls in the first 4 and last 8 bases of the read
--invert-edge-filter
Invert the edge filter, instead of filtering out base modification
calls at the ends of reads, only _keep_ base modification calls at the
ends of reads. E.g. if usually, "4,8" would remove (i.e. filter out)
base modification calls in the first 4 and last 8 bases of the read,
using this flag will keep only base modification calls in the first 4
and last 8 bases
--filter-probs
Filter out the lowest confidence base modification probabilities
--mapped-only
Only use base modification probabilities from bases that are aligned
when estimating the filter threshold (i.e. ignore soft-clipped, and
inserted bases)
Sampling Options:
-n, --num-reads <NUM_READS>
Sample approximately this many reads when estimating the filtering
threshold. If alignments are present reads will be sampled evenly
across aligned genome. If a region is specified, either with the
--region option or the --sample-region option, then reads will be
sampled evenly across the region given. This option is useful for
large BAM files. In practice, 10-50 thousand reads is sufficient to
estimate the model output distribution and determine the filtering
threshold
[default: 10042]
--sample-region <SAMPLE_REGION>
Specify a region for sampling reads from when estimating the threshold
probability. If this option is not provided, but --region is provided,
the genomic interval passed to --region will be used. Format should be
<chrom_name>:<start>-<end> or <chrom_name>
--sampling-interval-size <SAMPLING_INTERVAL_SIZE>
Interval chunk size to process concurrently when estimating the
threshold probability, can be larger than the pileup processing
interval
[default: 1000000]
Filtering Options:
-p, --filter-percentile <FILTER_PERCENTILE>
Filter out modified base calls where the probability of the predicted
variant is below this confidence percentile. For example, 0.1 will
filter out the 10% lowest confidence modification calls
[default: 0.1]
--filter-threshold <FILTER_THRESHOLD>
Specify the filter threshold globally or per primary base. A global
filter threshold can be specified with by a decimal number (e.g.
0.75). Per-base thresholds can be specified by colon-separated values,
for example C:0.75 specifies a threshold value of 0.75 for cytosine
modification calls. Additional per-base thresholds can be specified by
repeating the option: for example --filter-threshold C:0.75
--filter-threshold A:0.70 or specify a single base option and a
default for all other bases with: --filter-threshold A:0.70
--filter-threshold 0.9 will specify a threshold value of 0.70 for
adenine and 0.9 for all other base modification calls
--mod-threshold <MOD_THRESHOLDS>
Specify a passing threshold to use for a base modification,
independent of the threshold for the primary sequence base or the
default. For example, to set the pass threshold for 5hmC to 0.8 use
`--mod-threshold h:0.8`. The pass threshold will still be estimated as
usual and used for canonical cytosine and other modifications unless
the `--filter-threshold` option is also passed. See the online
documentation for more details
Logging Options:
--suppress-progress
Hide the progress bar
update-tags
Renames Mm/Ml to tags to MM/ML. Also allows changing the mode flag from silent
'.' to explicitly '?' or '.'
Usage: modkit update-tags [OPTIONS] <IN_BAM> <OUT_BAM>
Arguments:
<IN_BAM> BAM to update modified base tags in. Can be a path to a file or one
of `-` or `stdin` to specify a stream from standard input
<OUT_BAM> File to new BAM file to be created or one of `-` or `stdin` to
specify a stream from standard output
Options:
-m, --mode <MODE> Mode, change mode to this value, options {'explicit',
'implicit'}. See spec at:
https://samtools.github.io/hts-specs/SAMtags.pdf.
'explicit' ('?') means residues without modification
probabilities will not be assumed canonical or
modified. 'implicit' means residues without explicit
modification probabilities are assumed to be
canonical [possible values: explicit, implicit]
--no-implicit-probs Don't add implicit canonical calls. This flag is
important when converting from one of the implicit
modes ( `.` or `""`) to explicit mode (`?`). By
passing this flag, the bases without associated base
modification probabilities will not be assumed to be
canonical. No base modification probability will be
written for these bases, meaning there is no
information. The mode will automatically be set to
the explicit mode `?`
-h, --help Print help
Compute Options:
-t, --threads <THREADS> Number of threads to use [default: 4]
Logging Options:
--log-filepath <LOG_FILEPATH> Output debug logs to file at this path
Output Options:
--output-sam Output SAM format instead of BAM
sample-probs
Calculate an estimate of the base modification probability distribution
Usage: modkit sample-probs [OPTIONS] <IN_BAM>
Arguments:
<IN_BAM>
Input BAM with modified base tags. If a index is found reads will be
sampled evenly across the length of the reference sequence. Can be a
path to a file or one of `-` or `stdin` to specify a stream from
standard input
Options:
--ignore-index
Ignore the BAM index (if it exists) perform a serial scan of the BAM,
otherwise if an index is found, multiple workers will be used to read
the BAM in parallel. Conflicts with --threads since there will only be
one reader
-r, --reference <REFERENCE_FASTA>
Reference sequence in FASTA format. (alias: 'ref')
--preload-references
Preload the reference sequences, useful when working with many, short
reference sequences such as a transcriptome
-k, --mask
Respect soft masking in the reference FASTA
-h, --help
Print help (see a summary with '-h')
Compute Options:
-t, --threads <THREADS>
Number of workers to run concurrently. When an aligned, indexed BAM is
found, multiple workers (threads) will be spawned to read disjoint
sections of the BAM in parallel
[default: 4]
--io-threads <IO_THREADS>
Number of IO/decompression threads to use, these are shared across
workers (if an indexed BAM is found) or used for a single reader if
--ignore-index is passed, stdin is used, or an index is not found
[default: 4]
-i, --interval-size <INTERVAL_SIZE>
Interval chunk size in base pairs to process concurrently. Smaller
interval chunk sizes will use less memory but incur more overhead.
Only used when sampling probs from an indexed bam
[default: 1000000]
Logging Options:
--log-filepath <LOG_FILEPATH>
Specify a file for debug logs to be written to, otherwise ignore them.
Setting a file is recommended
--suppress-progress
Hide the progress bar
Output Options:
-p, --percentiles <PERCENTILES>
Percentiles to calculate, a space separated list of floats (for
example: "10,20,90")
[default: 10,50,90]
-q, --quantiles <QUANTILES>
Quantiles to calculate, a space separated list of floats (for example:
"0.1,0.2,0.9")
-o, --out-dir <OUT_DIR>
Directory to deposit result tables into. Required for model
probability histogram output
--prefix <PREFIX>
Label to prefix output files with
--force
Overwrite results if present
--hist
Output histogram of base modification prediction probabilities
--dna-color <PRIMARY_BASE_COLORS> <PRIMARY_BASE_COLORS>
Set colors of primary bases in histogram, should be RGB format, e.g.
"#0000FF" is defailt for canonical cytosine
--mod-color <MOD_BASE_COLORS> <MOD_BASE_COLORS>
Set colors of modified bases in histogram, should be RGB format, e.g.
"#FF00FF" is default for 5hmC
Selection Options:
--motif <MOTIF> <MOTIF>
Tabulate base modification probabilities at motifs (or single bases).
The first argument should be the sequence motif and the second
argument is the 0-based offset to the base to pileup base modification
counts for. For example: --motif CGCG 0 indicates to collect base
mofification probabilities for the first C on the top strand and the
last C (complement to G) on the bottom strand. For single bases,
--motif C 0 or --motif A 0 would restrict to probabilities on C or A
reference bases, respectively. This argument can be passed multiple
times
--edge-filter <EDGE_FILTER>
Discard base modification calls that are this many bases from the
start or the end of the read. Two comma-separated values may be
provided to asymmetrically filter out base modification calls from the
start and end of the reads. For example, 4,8 will filter out base
modification calls in the first 4 and last 8 bases of the read
--region <REGION>
Process only the specified region of the BAM when collecting
probabilities. Format should be <chrom_name>:<start>-<end> or
<chrom_name>
--include-bed <INCLUDE_BED>
Only sample base modification probabilities that are aligned to the
positions in this BED file. (alias: include-positions)
--use-non-primary
Use non-primary mappings, may cause double-counting of reads with
primary and one or more non-primary mappings. Requires MN tag to be
correct. See documentation for help on proper alignment
Sampling Options:
-n, --num-reads <NUM_READS>
-f, --sampling-frac <SAMPLING_FRAC>
Instead of using a defined number of reads, specify a fraction of
reads to sample, for example 0.1 will sample 1/10th of the reads
--no-sampling
No sampling, use all of the reads to calculate the filter thresholds
-s, --seed <SEED>
Random seed for deterministic running, the default is
non-deterministic, only used when no BAM index is provided
summary
Summarize the mod tags present in a BAM and get basic statistics. The default
output is a totals table (designated by '#' lines) and a modification calls
table. Descriptions of the columns can be found in the README
Usage: modkit summary [OPTIONS] <IN_BAM>
Arguments:
<IN_BAM>
Input modBam, can be a path to a file or one of `-` or `stdin` to
specify a stream from standard input
Options:
--ignore-index
Ignore the BAM index (if it exists) perform a serial scan of the BAM,
otherwise if an index is found, multiple workers will be used to read
the BAM in parallel. Conflicts with --threads since there will only be
one reader
-r, --reference <REFERENCE_FASTA>
Reference sequence in FASTA format. (alias: 'ref')
--preload-references
Preload the reference sequences, useful when working with many, short
reference sequences such as a transcriptome
-k, --mask
Respect soft masking in the reference FASTA
--cpg
Shorthand for --motif CG 0. Requires a sorted, indexed modBAM
-h, --help
Print help (see a summary with '-h')
Compute Options:
-t, --threads <THREADS>
Number of threads to use
[default: 4]
--io-threads <IO_THREADS>
Number of IO/decompression threads to use, these are shared across
workers (if an indexed BAM is found) or used for a single reader if
--ignore-index is passed, stdin is used, or an index is not found
[default: 4]
-i, --interval-size <INTERVAL_SIZE>
When using regions, interval chunk size in base pairs to process
concurrently. Smaller interval chunk sizes will use less memory but
incur more overhead
[default: 1000000]
Logging Options:
--log-filepath <LOG_FILEPATH>
Specify a file for debug logs to be written to, otherwise ignore them.
Setting a file is recommended
--suppress-progress
Hide the progress bar
Output Options:
--tsv
Output summary as a tab-separated variables stdout instead of a table
--combine-mods
Combine modification counts per primary sequence base when calculating
summary statistics
Sampling Options:
-n, --num-reads <NUM_READS>
Approximate maximum number of reads to use. If an indexed BAM is
provided, the reads will be sampled evenly over the length of the
aligned reference. If a region is passed with the --region option,
they will be sampled over the region. Actual number of reads used may
deviate slightly from this number
-f, --sampling-frac <SAMPLING_FRAC>
Instead of using a defined number of reads, specify a fraction of
reads to sample when estimating the filter threshold. For example 0.1
will sample 1/10th of the reads
-s, --seed <SEED>
Sets a random seed for deterministic running (when using
--sample-frac)
Filtering Options:
-p, --filter-quantile <FILTER_QUANTILE>
Filter out modified base calls where the probability of the predicted
variant is below this confidence percentile. For example, 0.1 will
filter out the 10% lowest confidence base modification calls
[default: 0.1]
--filter-threshold <FILTER_THRESHOLD>
Specify the filter threshold globally or per-base. Global filter
threshold can be specified with by a decimal number (e.g. 0.75).
Per-base thresholds can be specified by colon-separated values, for
example C:0.75 specifies a threshold value of 0.75 for cytosine
modification calls. Additional per-base thresholds can be specified by
repeating the option: for example --filter-threshold C:0.75
--filter-threshold A:0.70 or specify a single base option and a
default for all other bases with: --filter-threshold A:0.70
--filter-threshold 0.9 will specify a threshold value of 0.70 for
adenine and 0.9 for all other base modification calls
--mod-threshold <MOD_THRESHOLDS>
Specify a passing threshold to use for a base modification,
independent of the threshold for the primary sequence base or the
default. For example, to set the pass threshold for 5hmC to 0.8 use
`--mod-threshold h:0.8`. The pass threshold will still be estimated as
usual and used for canonical cytosine and other modifications unless
the `--filter-threshold` option is also passed. See the online
documentation for more details
--max-filter-threshold <MAX_FILTER_THRESHOLD>
Maximum threhsold value to use, if the estimated threshold value for
any primary sequence base is greater than this value, use this value
instead. To set a default value for all primary bases, use
--max-threshold 0.8, for example. Or use per-base threshold values
like --max-threshold C:0.8
Selection Options:
--edge-filter <EDGE_FILTER>
Discard base modification calls that are this many bases from the
start or the end of the read. Two comma-separated values may be
provided to asymmetrically filter out base modification calls from the
start and end of the reads. For example, 4,8 will filter out base
modification calls in the first 4 and last 8 bases of the read
--include-bed <INCLUDE_BED>
Only summarize base modification probabilities that are aligned to the
positions in this BED file. (alias: include-positions)
--mapped-only
Only use modBAM records that are mapped, don't include unmapped
records
--matched-only
Only summarize base modifications that are mapped and are matches to
the appropriate primary sequence base. For example, require that 5mC
base modification calls be aligned to a reference C. Requires a
reference FASTA and an sorted, indexed modBAM
--motif <MOTIF> <MOTIF>
Calculate base modification statistics at reference motifs. This
option has the same behavior as "--matched-only" except it only
includes specified motifs. The first argument should be the sequence
motif and the second argument is the 0-based offset to the base to
pileup base modification counts for. For example: --motif CGCG 0
indicates to collect base mofification probabilities for the first C
on the top strand and the last C (complement to G) on the bottom
strand. For single bases, --motif C 0 or --motif A 0 would restrict to
probabilities on C or A reference bases, respectively. This argument
can be passed multiple times. Requires a sorted, indexed modBAM
--region <REGION>
Process only the specified region of the BAM when collecting
probabilities. Format should be <chrom_name>:<start>-<end> or
<chrom_name>
call-mods
Call mods from a modbam, creates a new modbam with probabilities set to 100% if
a base modification is called or 0% if called canonical
Usage: modkit call-mods [OPTIONS] <IN_BAM> <OUT_BAM>
Arguments:
<IN_BAM>
Input BAM, may be sorted and have associated index available. Can be a
path to a file or one of `-` or `stdin` to specify a stream from
standard input
<OUT_BAM>
Output BAM, can be a path to a file or one of `-` or `stdin` to
specify a stream from standard input
Options:
--log-filepath <LOG_FILEPATH>
Specify a file for debug logs to be written to, otherwise ignore them.
Setting a file is recommended
--ff
Fast fail, stop processing at the first invalid sequence record.
Default behavior is to continue and report failed/skipped records at
the end
--suppress-progress
Hide the progress bar
-t, --threads <THREADS>
Number of threads to use while processing chunks concurrently
[default: 4]
-n, --num-reads <NUM_READS>
Sample approximately this many reads when estimating the filtering
threshold. If alignments are present reads will be sampled evenly
across aligned genome. If a region is specified, either with the
--region option or the --sample-region option, then reads will be
sampled evenly across the region given. This option is useful for
large BAM files. In practice, 10-50 thousand reads is sufficient to
estimate the model output distribution and determine the filtering
threshold
[default: 10042]
-f, --sampling-frac <SAMPLING_FRAC>
Sample this fraction of the reads when estimating the
filter-percentile. In practice, 50-100 thousand reads is sufficient to
estimate the model output distribution and determine the filtering
threshold. See filtering.md for details on filtering
--seed <SEED>
Set a random seed for deterministic running, the default is
non-deterministic, only used when no BAM index is provided
--sample-region <SAMPLE_REGION>
Specify a region for sampling reads from when estimating the threshold
probability. If this option is not provided, but --region is provided,
the genomic interval passed to --region will be used. Format should be
<chrom_name>:<start>-<end> or <chrom_name>
--sampling-interval-size <SAMPLING_INTERVAL_SIZE>
Interval chunk size to process concurrently when estimating the
threshold probability, can be larger than the pileup processing
interval
[default: 1000000]
-p, --filter-percentile <FILTER_PERCENTILE>
Filter out modified base calls where the probability of the predicted
variant is below this confidence percentile. For example, 0.1 will
filter out the 10% lowest confidence modification calls
[default: 0.1]
--filter-threshold <FILTER_THRESHOLD>
Specify the filter threshold globally or per primary base. A global
filter threshold can be specified with by a decimal number (e.g.
0.75). Per-base thresholds can be specified by colon-separated values,
for example C:0.75 specifies a threshold value of 0.75 for cytosine
modification calls. Additional per-base thresholds can be specified by
repeating the option: for example --filter-threshold C:0.75
--filter-threshold A:0.70 or specify a single base option and a
default for all other bases with: --filter-threshold A:0.70
--filter-threshold 0.9 will specify a threshold value of 0.70 for
adenine and 0.9 for all other base modification calls
--mod-threshold <MOD_THRESHOLDS>
Specify a passing threshold to use for a base modification,
independent of the threshold for the primary sequence base or the
default. For example, to set the pass threshold for 5hmC to 0.8 use
`--mod-threshold h:0.8`. The pass threshold will still be estimated as
usual and used for canonical cytosine and other modifications unless
the `--filter-threshold` option is also passed. See the online
documentation for more details
--no-filtering
Don't filter base modification calls, assign each base modification to
the highest probability prediction
--edge-filter <EDGE_FILTER>
Discard base modification calls that are this many bases from the
start or the end of the read. Two comma-separated values may be
provided to asymmetrically filter out base modification calls from the
start and end of the reads. For example, 4,8 will filter out base
modification calls in the first 4 and last 8 bases of the read
--invert-edge-filter
Invert the edge filter, instead of filtering out base modification
calls at the ends of reads, only _keep_ base modification calls at the
ends of reads. E.g. if usually, "4,8" would remove (i.e. filter out)
base modification calls in the first 4 and last 8 bases of the read,
using this flag will keep only base modification calls in the first 4
and last 8 bases
--motif <MOTIF> <MOTIF>
Filter out any base modification call that isn't part of a basecall
sequence motif This argument can be passed multiple times. Format is
<motif_sequence> <offset>. For example the argument to match CpG
dinucleotides is `--motif CG 0`, or to match CG[5mC]G the argument
would be `--motif CGCG 2`
--cpg
Shorthand for --motif CG 0
--discard-motifs
Discard base modification calls that match the provided motifs
(instead of keeping them)
--output-sam
Output SAM format instead of BAM
-h, --help
Print help (see a summary with '-h')
repair
Repair MM and ML tags in one bam with the correct tags from another. To use this
command, both modBAMs _must_ be sorted by read name. The "donor" modBAM's reads
must be a superset of the acceptor's reads. Extra reads in the donor are
allowed, and multiple reads with the same name (secondary, etc.) are allowed in
the acceptor. Reads with an empty SEQ field cannot be repaired and will be
rejected. Reads where there is an ambiguous alignment of the acceptor to the
donor will be rejected (and logged). See the full documentation for details
Usage: modkit repair [OPTIONS] --donor-bam <DONOR_BAM> --acceptor-bam <ACCEPTOR_BAM> --output-bam <OUTPUT_BAM>
Options:
-d, --donor-bam <DONOR_BAM>
Donor modBAM with original MM/ML tags. Must be sorted by read name
-a, --acceptor-bam <ACCEPTOR_BAM>
Acceptor modBAM with reads to have MM/ML base modification data
projected on to. Must be sorted by read name
-o, --output-bam <OUTPUT_BAM>
output modBAM location
--log-filepath <LOG_FILEPATH>
File to write logs to, it is recommended to use this option as some
reads may be rejected and logged here
-t, --threads <THREADS>
The number of threads to use [default: 4]
-h, --help
Print help
validate
Validate results from a set of mod-BAM files and associated BED files containing
the ground truth modified base status at reference positions
Usage: modkit validate [OPTIONS]
Options:
-h, --help
Print help (see a summary with '-h')
Sample Options:
--bam-and-bed <BAM> <BED>
Argument accepts 2 values. The first value is the BAM file path with
modified base tags. The second is a bed file with ground truth
reference positions. The name field in the ground truth bed file
should be the short name (single letter code or ChEBI ID) for a
modified base or `-` to specify a canonical base ground truth
position. This argument can be provided more than once for multiple
samples
-c, --canonical-base <CANONICAL_BASE>
Canonical base to evaluate. By default, this will be derived from mod
codes in ground truth BED files. For ground truth with only canonical
sites and/or ChEBI codes this values must be set
[possible values: A, C, G, T]
Modified Base Options:
--ignore <IGNORE>
Ignore a modified base class _in_situ_ by redistributing base
modification probability equally across other options. For example, if
collapsing 'h', with 'm' and canonical options, half of the
probability of 'h' will be added to both 'm' and 'C'. A full
description of the methods can be found in collapse.md
Selection Options:
--edge-filter <EDGE_FILTER>
Discard base modification calls that are this many bases from the
start or the end of the read. Two comma-separated values may be
provided to asymmetrically filter out base modification calls from the
start and end of the reads. For example, 4,8 will filter out base
modification calls in the first 4 and last 8 bases of the read
--invert-edge-filter
Invert the edge filter, instead of filtering out base modification
calls at the ends of reads, only _keep_ base modification calls at the
ends of reads. E.g. if usually, "4,8" would remove (i.e. filter out)
base modification calls in the first 4 and last 8 bases of the read,
using this flag will keep only base modification calls in the first 4
and last 8 bases
--min-identity <MIN_ALIGNMENT_IDENTITY>
Only use reads with alignment identity >= this number, in Q-space
(phred score)
--min-length <MIN_ALIGNMENT_LENGTH>
Remove reads with fewer aligned reference bases than this threshold
Filtering Options:
-p, --filter-quantile <FILTER_QUANTILE>
Filter out modified base calls where the probability of the predicted
variant is below this confidence percentile. For example, 0.1 will
filter out the 10% lowest confidence modification calls
[default: 0.1]
--filter-threshold <FILTER_THRESHOLD>
Specify modified base probability filter threshold value. If
specified, --filter-threshold will override --filter-quantile
Compute Options:
-t, --threads <THREADS>
Number of threads to use
[default: 4]
Logging Options:
--suppress-progress
Hide the progress bar
--log-filepath <LOG_FILEPATH>
Specify a file for debug logs to be written to, otherwise ignore them.
Setting a file is recommended. (alias: log)
Output Options:
-o, --out-filepath <OUT_FILEPATH>
Specify a file for machine parseable output
pileup-hemi
Tabulates double-stranded base modification patters (such as hemi-methylation)
across genomic motif positions. This command produces a bedMethyl file, the
schema can be found in the online documentation
Usage: modkit pileup-hemi [OPTIONS] --ref <REFERENCE_FASTA> <IN_BAM>
Arguments:
<IN_BAM>
Input BAM, should be sorted and have associated index available
Options:
-o, --out-bed <OUT_BED>
Output file to write results into. Will write to stdout if not
provided
-h, --help
Print help (see a summary with '-h')
Modified Base Options:
--cpg
Aggregate double-stranded base modifications for CpG dinucleotides.
This flag is short-hand for --motif CG 0
--motif <MOTIF> <MOTIF>
Specify the sequence motif to pileup double-stranded base modification
pattern counts for. The first argument should be the sequence motif
and the second argument is the 0-based offset to the base to pileup
base modification counts for. For example: --motif CG 0 indicates to
generate pattern counts for the C on the top strand and the following
C (opposite to G) on the negative strand. The motif must be
reverse-complement palindromic or an error will be raised. See the
documentation for more examples and details
-r, --ref <REFERENCE_FASTA>
Reference sequence in FASTA format
--ignore <IGNORE>
Ignore a modified base class _in_situ_ by redistributing base
modification probability equally across other options. For example, if
collapsing 'h', with 'm' and canonical options, half of the
probability of 'h' will be added to both 'm' and 'C'. A full
description of the methods can be found in collapse.md
--force-allow-implicit
Force allow implicit-canonical mode. By default modkit does not allow
pileup with the implicit mode (e.g. C+m, no '.' or '?'). The
`update-tags` subcommand is provided to update tags to the new mode.
This option allows the interpretation of implicit mode tags: residues
without modified base probability will be interpreted as being the
non-modified base
-k, --mask
Respect soft masking in the reference FASTA
--combine-mods
Combine base modification calls, all counts of modified bases are
summed together. See collapse.md for details
Logging Options:
--log-filepath <LOG_FILEPATH>
Specify a file for debug logs to be written to, otherwise ignore them.
Setting a file is recommended. (alias: log)
--suppress-progress
Hide the progress bar
Selection Options:
--region <REGION>
Process only the specified region of the BAM when performing pileup.
Format should be <chrom_name>:<start>-<end> or <chrom_name>. Commas
are allowed
--max-depth <MAX_DEPTH>
Maximum number of records to use when calculating pileup. This
argument is passed to the pileup engine. If you have high depth data,
consider increasing this value substantially. Must be less than
2147483647 or an error will be raised
[default: 8000]
--include-bed <INCLUDE_BED>
BED file that will restrict threshold estimation and pileup results to
positions overlapping intervals in the file. (alias:
include-positions)
--include-unmapped
Include unmapped base modifications when estimating the pass threshold
--edge-filter <EDGE_FILTER>
Discard base modification calls that are this many bases from the
start or the end of the read. Two comma-separated values may be
provided to asymmetrically filter out base modification calls from the
start and end of the reads. For example, 4,8 will filter out base
modification calls in the first 4 and last 8 bases of the read
--invert-edge-filter
Invert the edge filter, instead of filtering out base modification
calls at the ends of reads, only _keep_ base modification calls at the
ends of reads. E.g. if usually, "4,8" would remove (i.e. filter out)
base modification calls in the first 4 and last 8 bases of the read,
using this flag will keep only base modification calls in the first 4
and last 8 bases
Compute Options:
-t, --threads <THREADS>
Number of threads to use while processing chunks concurrently
[default: 4]
-i, --interval-size <INTERVAL_SIZE>
Interval chunk size in base pairs to process concurrently. Smaller
interval chunk sizes will use less memory but incur more overhead
[default: 100000]
--queue-size <QUEUE_SIZE>
Size of queue for writing records
[default: 1000]
--chunk-size <CHUNK_SIZE>
Break contigs into chunks containing this many intervals (see
`interval_size`). This option can be used to help prevent excessive
memory usage, usually with no performance penalty. By default, modkit
will set this value to 1.5x the number of threads specified, so if 4
threads are specified the chunk_size will be 6. A warning will be
shown if this option is less than the number of threads specified
Sampling Options:
-n, --num-reads <NUM_READS>
Sample this many reads when estimating the filtering threshold. Reads
will be sampled evenly across aligned genome. If a region is
specified, either with the --region option or the --sample-region
option, then reads will be sampled evenly across the region given.
This option is useful for large BAM files. In practice, 10-50 thousand
reads is sufficient to estimate the model output distribution and
determine the filtering threshold
[default: 10042]
-f, --sampling-frac <SAMPLING_FRAC>
Sample this fraction of the reads when estimating the
filter-percentile. In practice, 50-100 thousand reads is sufficient to
estimate the model output distribution and determine the filtering
threshold. See filtering.md for details on filtering
--seed <SEED>
Set a random seed for deterministic running, the default is
non-deterministic
Filtering Options:
--no-filtering
Do not perform any filtering, include all mod base calls in output.
See filtering.md for details on filtering
-p, --filter-percentile <FILTER_PERCENTILE>
Filter out modified base calls where the probability of the predicted
variant is below this confidence percentile. For example, 0.1 will
filter out the 10% lowest confidence modification calls
[default: 0.1]
--filter-threshold <FILTER_THRESHOLD>
Specify the filter threshold globally or per-base. Global filter
threshold can be specified with by a decimal number (e.g. 0.75).
Per-base thresholds can be specified by colon-separated values, for
example C:0.75 specifies a threshold value of 0.75 for cytosine
modification calls. Additional per-base thresholds can be specified by
repeating the option: for example --filter-threshold C:0.75
--filter-threshold A:0.70 or specify a single base option and a
default for all other bases with: --filter-threshold A:0.70
--filter-threshold 0.9 will specify a threshold value of 0.70 for
adenine and 0.9 for all other base modification calls
--mod-threshold <MOD_THRESHOLDS>
Specify a passing threshold to use for a base modification,
independent of the threshold for the primary sequence base or the
default. For example, to set the pass threshold for 5hmC to 0.8 use
`--mod-threshold h:0.8`. The pass threshold will still be estimated as
usual and used for canonical cytosine and other modifications unless
the `--filter-threshold` option is also passed. See the online
documentation for more details
--sample-region <SAMPLE_REGION>
Specify a region for sampling reads from when estimating the threshold
probability. If this option is not provided, but --region is provided,
the genomic interval passed to --region will be used. Format should be
<chrom_name>:<start>-<end> or <chrom_name>
--sampling-interval-size <SAMPLING_INTERVAL_SIZE>
Interval chunk size in base pairs to process concurrently when
estimating the threshold probability, can be larger than the pileup
processing interval
[default: 1000000]
Output Options:
--only-tabs
**Deprecated** The default output has all tab-delimiters. For
bedMethyl output, separate columns with only tabs. The default is to
use tabs for the first 10 fields and spaces thereafter. The default
behavior is more likely to be compatible with genome viewers. Enabling
this option may make it easier to parse the output with tabular data
handlers that expect a single kind of separator
--mixed-delim
Output bedMethyl where the delimiter of columns past column 10 are
space-delimited instead of tab-delimited. This option can be useful
for some browsers and parsers that don't expect the extra columns of
the bedMethyl format
entropy
Use a mod-BAM to calculate methylation entropy over genomic windows
Usage: modkit entropy [OPTIONS] --in-bam <IN_BAMS> --ref <REFERENCE_FASTA>
Options:
-s, --in-bam <IN_BAMS>
Input mod-BAM, may be repeated multiple times to calculate entropy
across all input mod-BAMs
-n, --num-positions <NUM_POSITIONS>
Number of modified positions to consider at a time
[default: 4]
-w, --window-size <WINDOW_SIZE>
Maximum length interval that "num_positions" modified bases can occur
in. The maximum window size decides how dense the positions are
packed. For example, consider that the num_positions is equal to 4,
the motif is CpG, and the window_size is equal to 8, this
configuration would require that the modified positions are
immediately adjacent to each other, "CGCGCGCG". On the other hand, if
the window_size was set to 12, then multiple sequences with various
patterns of other bases can be used CGACGATCGGCG
[default: 50]
--ref <REFERENCE_FASTA>
Reference sequence in FASTA format
--mask
Respect soft masking in the reference FASTA
--motif <MOTIF> <MOTIF>
Motif to use for entropy calculation, multiple motifs can be used by
repeating this option. When multiple motifs are used that specify
different modified primary bases, all modification possibilities will
be used in the calculation
--cpg
Use CpG motifs. Short hand for --motif CG 0 --combine-strands
--base <BASE>
Primary sequence base to calculate modification entropy on
[possible values: A, C, G, T]
--regions <REGIONS_FP>
Regions over which to calculate descriptive statistics
--combine-strands
Combine modification counts on the positive and negative strands and
report entropy on just the positive strand
--min-coverage <MIN_VALID_COVERAGE>
Minimum coverage required at each position in the window. Windows
without at least this many valid reads will be skipped, but positions
within the window with enough coverage can be used by neighboring
windows
[default: 3]
--max-filtered-positions <MAX_FILTERED_POSITIONS>
Maximum number of filtered positions a read is allowed to have in a
window, more than this number and the read will be discarded. Default
will be 50% of `num_positions`
-h, --help
Print help (see a summary with '-h')
Output Options:
-o, --out-bed <OUT_BED>
Output BED file, if using `--region` this must be a directory
--prefix <PREFIX>
Only used with `--regions`, prefix files in output directory with this
string
--force
Force overwrite output
--header
Write a header line
--drop-zeros
Omit windows with zero entropy
Filtering Options:
--no-filtering
Do not perform any filtering, include all mod base calls in output
-p, --filter-percentile <FILTER_PERCENTILE>
Filter out modified base calls where the probability of the predicted
variant is below this confidence percentile. For example, 0.1 will
filter out the 10% lowest confidence modification calls
[default: 0.1]
--filter-threshold <FILTER_THRESHOLD>
Specify the filter threshold globally or for the canonical calls. When
specified, base modification call probabilities will be required to be
greater than or equal to this number. If `--mod-threshold` is also
specified, _this_ value will be used for canonical calls
--mod-threshold <MOD_THRESHOLDS>
Specify a passing threshold to use for a base modification,
independent of the threshold for the primary sequence base or the
default. For example, to set the pass threshold for 5hmC to 0.8 use
`--mod-threshold h:0.8`. The pass threshold will still be estimated as
usual and used for canonical cytosine and other modifications unless
the `--filter-threshold` option is also passed. See the online
documentation for more details
Sampling Options:
--num-reads <NUM_READS>
Sample this many reads when estimating the filtering threshold. Reads
will be sampled evenly across aligned genome. If a region is
specified, either with the --region option or the --sample-region
option, then reads will be sampled evenly across the region given.
This option is useful for large BAM files. In practice, 10-50 thousand
reads is sufficient to estimate the model output distribution and
determine the filtering threshold
[default: 10042]
Compute Options:
-t, --threads <THREADS>
Number of threads to use
[default: 4]
--io-threads <IO_THREADS>
Number of BAM-reading threads to use
Logging Options:
--log-filepath <LOG_FILEPATH>
Send debug logs to this file, setting this file is recommended
--verbose-logging
Log regions that have zero or insufficient coverage. Requires log file
--suppress-progress
Hide progress bars
localize
Investigate patterns of base modifications, by aggregating pileup counts
"localized" around genomic features of interest
Usage: modkit localize [OPTIONS] --regions <REGIONS> --genome-sizes <GENOME_SIZES> <IN_BEDMETHYL>
Arguments:
<IN_BEDMETHYL>
Input bedMethyl table. Should be bgzip-compressed and have an
associated Tabix index. The tabix index will be assumed to be
$this_file.tbi
Options:
--regions <REGIONS>
BED file of regions to calculate enrichment around. These BED records
serve as the points from which the `--window` number of bases is
centered
-w, --window <EXPAND_WINDOW>
Number of base pairs to search around, for example if your BED region
records are single positions, a window of 500 will look 500 base pairs
upstream and downstream of that position. If your region BED records
are larger regions, this will expand from the midpoint of that region
[default: 2000]
-s, --stranded <STRANDED>
Whether to only keep bedMethyl records on the "same" strand or
"opposite" strand
[possible values: same, opposite]
--stranded-features <STRANDED_FEATURES>
Force use bedMethyl records from a particular strand, default is to
use the strand as given in the BED file (will use BOTH for BED3)
[possible values: positive, negative, both]
--min-coverage <MIN_COVERAGE>
Minimum valid coverage to use a bedMethyl record
[default: 3]
-r, --genome-sizes <GENOME_SIZES>
TSV of genome sizes, should be <chrom>\t<size_in_bp>
-o, --out-file <OUT_FILE>
Optionally specify a file to write output to, default is stdout
-h, --help
Print help (see a summary with '-h')
Output Options:
--chart <CHART_FILEPATH>
Create plots showing %-modification vs. offset. Argument should be a
path to a file
--name <CHART_NAME>
Give the HTML document and chart a name
-f, --force
Force overwrite of existing output file
Logging Options:
--log-filepath <LOG_FILEPATH>
Specify a file to write debug logs to
Compute Options:
-t, --threads <THREADS>
Number of threads to use
[default: 4]
--io-threads <IO_THREADS>
Number of tabix/bgzf IO threads to use
[default: 2]
--batch-size <BATCH_SIZE_BP>
[default: 500000]
stats
Calculate base modification levels over regions
Usage: modkit stats [OPTIONS] --regions <REGIONS> --out-table <OUT_TABLE> <IN_BEDMETHYL>
Arguments:
<IN_BEDMETHYL> Input bedMethyl table. Should be bgzip-compressed and have an
associated Tabix index. The tabix index will be assumed to be
$this_file.tbi
Options:
--regions <REGIONS>
BED file of regions to aggregate base modification over
-c, --mod-codes <MOD_CODES>
Specify which base modification codes to use. Default will report
information on all base modification codes encountered
-m, --min-coverage <MIN_COVERAGE>
Only use records with at least this much valid coverage [default: 1]
-h, --help
Print help
Output Options:
-o, --out-table <OUT_TABLE> Specify the output file to write the results
table, or "-"/"stdout" for stdout
--force Force overwrite the output file
--no-header Don't add the header describing the columns to
the output
Logging Options:
--log-filepath <LOG_FILEPATH> Specify a file to write debug logs to
Compute Options:
-t, --threads <THREADS> Number of threads to use [default: 4]
--io-threads <IO_THREADS> Number of tabix/bgzf threads to use [default:
2]
open-chromatin predict
Usage: modkit open-chromatin predict [OPTIONS] --model <MODEL_PATH> --output <OUTPUT> <IN_BAM>
Arguments:
<IN_BAM>
Input modBAM with 6mA base modification calls
Options:
--model <MODEL_PATH>
Path to directory with open-chromatin model
--batch-size <BATCH_SIZE>
Collect this many windows of data for each run through the model
[default: 1024]
--super-batch-size <SUPER_BATCH_SIZE>
Number of "batches" to collect at once, see documentation for exact
calculation or run with --dryrun to see output
[default: 100]
--step-size <STEP_SIZE>
Number of base pairs to step along the genome or genomic intervals to
make predictions. Smaller numbers will result in better resolution but
take more computation
[default: 25]
-t, --min-coverage <MIN_COVERAGE>
Require this many reads covering each prediction window
[default: 5]
--threshold <FILTER_THRESHOLD>
Only emit records/windows where the open chromatin probability is
greater than or equal to this value
-o, --output <OUTPUT>
Output bedGraph file, or "-"/"stdout" to write to stdout
--force
Force overwrite output file
--log-filepath <LOG_FILEPATH>
Path to file to write logs to, setting this parameter is recommended
--include-bed <INCLUDE_BED>
BED file of regions over which to make predictions
--region <REGION>
Region in the format <chrom>:start-end over which to make predictions
--suppress-progress
Hide the progress bar
--no-header
Don't print header in output
--dryrun
Perform setup and validations, but stop before performing inference
-h, --help
Print help (see a summary with '-h')
extract full
Transform the probabilities from the MM/ML tags in a modBAM into a table
Usage: modkit extract full [OPTIONS] <IN_BAM> <OUT_PATH>
Arguments:
<IN_BAM>
Path to modBAM file to extract read-level information from, or one of
`-` or `stdin` to specify a stream from standard input. If a file is
used it may be sorted and have associated index
<OUT_PATH>
Path to output file, "stdout" or "-" will direct output to standard
out
Options:
--reference <REFERENCE>
Path to reference FASTA to extract reference context information from.
Required for motif selection
-h, --help
Print help (see a summary with '-h')
Compute Options:
-t, --threads <THREADS>
Number of threads to use
[default: 4]
--io-threads <IO_THREADS>
Number of IO/decompression threads to use
[default: 4]
--out-threads <OUT_THREADS>
Number of threads to use for parallel bgzf writing
[default: 4]
-q, --queue-size <QUEUE_SIZE>
Number of reads that can be in memory at a time. Increasing this value
will increase thread usage, at the cost of memory usage
[default: 10000]
--ignore-index
Ignore the BAM index (if it exists) and default to a serial scan of
the BAM
-i, --interval-size <INTERVAL_SIZE>
Interval chunk size in base pairs to process concurrently. Smaller
interval chunk sizes will use less memory but incur more overhead.
Only used when an indexed modBAM is provided
[default: 100000]
Output Options:
--bgzf
Write output as BGZF compressed file
--force
Force overwrite of output file
--kmer-size <KMER_SIZE>
Set the query and reference k-mer size (if a reference is provided).
Maximum number for this value is 50
[default: 5]
--no-headers
Don't print the header lines in the output tables
Logging Options:
--log-filepath <LOG_FILEPATH>
Path to file to write run log
--suppress-progress
Hide the progress bar
Selection Options:
--mapped-only
Include only mapped bases in output (alias: mapped)
--allow-non-primary
Output aligned secondary and supplementary base modification
probabilities as additional rows. The primary alignment will have all
of the base modification probabilities (including soft-clipped ones,
unless --mapped-only is used). The non-primary alignments will only
have mapped bases in the output
--num-reads <NUM_READS>
Number of reads to use. Note that when using a sorted, indexed modBAM
that the sampling algorithm will attempt to sample records evenly over
the length of the reference sequence. The result is the final number
of records used may be slightly more or less than the requested
number. When piping from stdin or using a modBAM without an index, the
requested number of reads will be the first `num_reads` records
--region <REGION>
Process only reads that are aligned to a specified region of the BAM.
Format should be <chrom_name>:<start>-<end> or <chrom_name>
--include-bed <INCLUDE_BED>
BED file with regions to include (alias: include-positions).
Implicitly only includes mapped sites
-v, --exclude-bed <EXCLUDE_BED>
BED file with regions to _exclude_ (alias: exclude)
--edge-filter <EDGE_FILTER>
Discard base modification calls that are this many bases from the
start or the end of the read. Two comma-separated values may be
provided to asymmetrically filter out base modification calls from the
start and end of the reads. For example, 4,8 will filter out base
modification calls in the first 4 and last 8 bases of the read
--invert-edge-filter
Invert the edge filter, instead of filtering out base modification
calls at the ends of reads, only _keep_ base modification calls at the
ends of reads. E.g. if usually, "4,8" would remove (i.e. filter out)
base modification calls in the first 4 and last 8 bases of the read,
using this flag will keep only base modification calls in the first 4
and last 8 bases
--ignore-implicit
Ignore implicitly canonical base modification calls. When the `.` flag
is used in the MM tag, this implies that bases missing a base
modification probability are to be assumed canonical. Set this flag to
omit those base modifications from the output. For additional details
see the SAM spec: https://samtools.github.io/hts-specs/SAMtags.pdf
Modified Base Options:
--motif <MOTIF> <MOTIF>
Output read-level base modification probabilities restricted to the
reference sequence motifs provided. The first argument should be the
sequence motif and the second argument is the 0-based offset to the
base to pileup base modification counts for. For example: --motif CGCG
0 indicates include base modifications for which the read is aligned
to the first C on the top strand and the last C (complement to G) on
the bottom strand. The --cpg argument is short hand for --motif CG 0.
This argument can be passed multiple times
--annotate-motifs
When used with `--motif` or `--cpg` emit all modified base alignment
information even if it does not align to a reference motif, but
annotate which aligned positions match which motifs in the "motifs"
column. "." will be used when an aligned position does not match a
motif
--cpg
Only output counts at CpG motifs. Requires a reference sequence to be
provided
-k, --mask
When using motifs, respect soft masking in the reference sequence
--ignore <IGNORE>
Ignore a modified base class _in_situ_ by redistributing base
modification probability equally across other options. For example, if
collapsing 'h', with 'm' and canonical options, half of the
probability of 'h' will be added to both 'm' and 'C'. A full
description of the methods can be found in collapse.md
extract calls
Produce a table of read-level base modification calls. This table has, for each
read, one row for each base modification call in that read using the same
thresholding algorithm as in pileup, or summary (see online documentation for
details on thresholds)
Usage: modkit extract calls [OPTIONS] <IN_BAM> <OUT_PATH>
Arguments:
<IN_BAM>
Path to modBAM file to extract read-level information from, or one of
`-` or `stdin` to specify a stream from standard input. If a file is
used it may be sorted and have associated index
<OUT_PATH>
Path to output file, "stdout" or "-" will direct output to standard
out
Options:
--reference <REFERENCE>
Path to reference FASTA to extract reference context information from.
If no reference is provided, `ref_kmer` column will be "." in the
output. (alias: ref)
--preload-references
Preload the reference sequences, useful when working with many, short
reference sequences such as a transcriptome
-h, --help
Print help (see a summary with '-h')
Compute Options:
-t, --threads <THREADS>
Number of threads to use
[default: 4]
--io-threads <IO_THREADS>
Number of IO/decompression threads to use
[default: 4]
--out-threads <OUT_THREADS>
Number of threads to use for parallel bgzf writing
[default: 4]
-q, --queue-size <QUEUE_SIZE>
Number of reads that can be in memory at a time. Increasing this value
will increase thread usage, at the cost of memory usage
[default: 10000]
--ignore-index
Ignore the BAM index (if it exists) and default to a serial scan of
the BAM
-i, --interval-size <INTERVAL_SIZE>
Interval chunk size in base pairs to process concurrently. Smaller
interval chunk sizes will use less memory but incur more overhead.
Only used when an indexed modBAM is provided
[default: 100000]
Output Options:
--bgzf
Write output as BGZF compressed file
--force
Force overwrite of output file
--kmer-size <KMER_SIZE>
Set the query and reference k-mer size (if a reference is provided).
Maximum number for this value is 50
[default: 5]
--no-headers
Don't print the header lines in the output tables
Logging Options:
--log-filepath <LOG_FILEPATH>
Path to file to write run log
--suppress-progress
Hide the progress bar
Selection Options:
--mapped-only
Include only mapped bases in output (alias: mapped)
--allow-non-primary
Output aligned secondary and supplementary base modification
probabilities as additional rows. The primary alignment will have all
of the base modification probabilities (including soft-clipped ones,
unless --mapped-only is used). The non-primary alignments will only
have mapped bases in the output
--num-reads <NUM_READS>
Number of reads to use. Note that when using a sorted, indexed modBAM
that the sampling algorithm will attempt to sample records evenly over
the length of the reference sequence. The result is the final number
of records used may be slightly more or less than the requested
number. When piping from stdin or using a modBAM without an index, the
requested number of reads will be the first `num_reads` records
--region <REGION>
Process only reads that are aligned to a specified region of the BAM.
Format should be <chrom_name>:<start>-<end> or <chrom_name>
--include-bed <INCLUDE_BED>
BED file with regions to include (alias: include-positions).
Implicitly only includes mapped sites
-v, --exclude-bed <EXCLUDE_BED>
BED file with regions to _exclude_ (alias: exclude)
--edge-filter <EDGE_FILTER>
Discard base modification calls that are this many bases from the
start or the end of the read. Two comma-separated values may be
provided to asymmetrically filter out base modification calls from the
start and end of the reads. For example, 4,8 will filter out base
modification calls in the first 4 and last 8 bases of the read
--invert-edge-filter
Invert the edge filter, instead of filtering out base modification
calls at the ends of reads, only _keep_ base modification calls at the
ends of reads. E.g. if usually, "4,8" would remove (i.e. filter out)
base modification calls in the first 4 and last 8 bases of the read,
using this flag will keep only base modification calls in the first 4
and last 8 bases
--ignore-implicit
Ignore implicitly canonical base modification calls. When the `.` flag
is used in the MM tag, this implies that bases missing a base
modification probability are to be assumed canonical. Set this flag to
omit those base modifications from the output. For additional details
see the SAM spec: https://samtools.github.io/hts-specs/SAMtags.pdf
--pass-only
Only output base modification calls that pass the minimum confidence
threshold. (alias: pass)
Modified Base Options:
--motif <MOTIF> <MOTIF>
Output read-level base modification probabilities restricted to the
reference sequence motifs provided. The first argument should be the
sequence motif and the second argument is the 0-based offset to the
base to pileup base modification counts for. For example: --motif CGCG
0 indicates include base modifications for which the read is aligned
to the first C on the top strand and the last C (complement to G) on
the bottom strand. The --cpg argument is short hand for --motif CG 0.
This argument can be passed multiple times
--annotate-motifs
When used with `--motif` or `--cpg` emit all modified base alignment
information even if it does not align to a reference motif, but
annotate which aligned positions match which motifs in the "motifs"
column. "." will be used when an aligned position does not match a
motif
--cpg
Only output counts at CpG motifs. Requires a reference sequence to be
provided
-k, --mask
When using motifs, respect soft masking in the reference sequence
--ignore <IGNORE>
Ignore a modified base class _in_situ_ by redistributing base
modification probability equally across other options. For example, if
collapsing 'h', with 'm' and canonical options, half of the
probability of 'h' will be added to both 'm' and 'C'. A full
description of the methods can be found in collapse.md
Filtering Options:
--filter-threshold <FILTER_THRESHOLD>
Specify the filter threshold globally or per-base. Global filter
threshold can be specified with by a decimal number (e.g. 0.75).
Per-base thresholds can be specified by colon-separated values, for
example C:0.75 specifies a threshold value of 0.75 for cytosine
modification calls. Additional per-base thresholds can be specified by
repeating the option: for example --filter-threshold C:0.75
--filter-threshold A:0.70 or specify a single base option and a
default for all other bases with: --filter-threshold A:0.70
--filter-threshold 0.9 will specify a threshold value of 0.70 for
adenine and 0.9 for all other base modification calls
--mod-threshold <MOD_THRESHOLDS>
Specify a passing threshold to use for a base modification,
independent of the threshold for the primary sequence base or the
default. For example, to set the pass threshold for 5hmC to 0.8 use
`--mod-threshold h:0.8`. The pass threshold will still be estimated as
usual and used for canonical cytosine and other modifications unless
the `--filter-threshold` option is also passed. See the online
documentation for more details
--max-filter-threshold <MAX_FILTER_THRESHOLD>
Maximum threhsold value to use, if the estimated threshold value for
any primary sequence base is greater than this value, use this value
instead. To set a default value for all primary bases, use
--max-threshold 0.8, for example. Or use per-base threshold values
like --max-threshold C:0.8
--no-filtering
Don't estimate the pass threshold, all calls will "pass"
-p, --filter-percentile <FILTER_PERCENTILE>
Filter out modified base calls where the probability of the predicted
variant is below this confidence percentile. For example, 0.1 will
filter out the 10% lowest confidence modification calls
[default: 0.1]
Sampling Options:
--sampling-interval-size <SAMPLING_INTERVAL_SIZE>
Interval chunk size in base pairs to process concurrently when
estimating the threshold probability
[default: 1000000]
-f, --sampling-frac <SAMPLING_FRAC>
Sample this fraction of the reads when estimating the pass-threshold.
In practice, 10-100 thousand reads is sufficient to estimate the model
output distribution and determine the filtering threshold. See
filtering.md for details on filtering
-n, --sample-num-reads <SAMPLE_NUM_READS>
Sample this many reads when estimating the filtering threshold. If a
sorted, indexed modBAM is provided reads will be sampled evenly across
aligned genome. If a region is specified, with the --region, then
reads will be sampled evenly across the region given. This option is
useful for large BAM files. In practice, 10-50 thousand reads is
sufficient to estimate the model output distribution and determine the
filtering threshold
[default: 10042]
--seed <SEED>
Set a random seed for deterministic running, the default is
non-deterministic when using `sampling_frac`. When using `num_reads`
the output is still deterministic
extract read-stats
Produce a table where modification counts are summarized on the read level. This
table will have one record per valid read and count the number of modified and
unmodified bases for each base modification requested
Usage: modkit extract read-stats [OPTIONS] <IN_BAM> <OUT_PATH>
Arguments:
<IN_BAM>
Input modBAM. Can be a path to a file or "-"/"stdin" for standard
input
<OUT_PATH>
Path to file for output, default will be to stdout
Options:
--filter-threshold <FILTER_THRESHOLD>
Specify the filter threshold globally or per-base. Global filter
threshold can be specified with by a decimal number (e.g. 0.75).
Per-base thresholds can be specified by colon-separated values, for
example C:0.75 specifies a threshold value of 0.75 for cytosine
modification calls. Additional per-base thresholds can be specified by
repeating the option: for example --filter-threshold C:0.75
--filter-threshold A:0.70 or specify a single base option and a
default for all other bases with: --filter-threshold A:0.70
--filter-threshold 0.9 will specify a threshold value of 0.70 for
adenine and 0.9 for all other base modification calls
--mod-thresholds <MOD_THRESHOLDS>
Specify a passing threshold to use for a base modification,
independent of the threshold for the primary sequence base or the
default. For example, to set the pass threshold for 5hmC to 0.8 use
`--mod-threshold h:0.8`. The pass threshold will still be estimated as
usual and used for canonical cytosine and other modifications unless
the `--filter-threshold` option is also passed
--mod-codes <MOD_CODES>...
Specify which modcodes to tabulate, should be
<primary_base>:<mod_code>
--queue-size <QUEUE_SIZE>
Queue size, maximum number of reads to be held in memory waiting for
records to be written
[default: 10000]
--io-threads <IO_THREADS>
[default: 4]
-h, --help
Print help (see a summary with '-h')
Logging Options:
--log-filepath <LOG_FILEPATH>
Path to file to write run log
--suppress-progress
Hide the progress bar
motif bed
Create BED file with all locations of a sequence motif. Example: modkit motif
bed CG 0
Usage: modkit motif bed [OPTIONS] <FASTA> <MOTIF> <OFFSET>
Arguments:
<FASTA> Input FASTA file
<MOTIF> Motif to search for within FASTA, e.g. CG
<OFFSET> Offset within motif, e.g. 0
Options:
-k, --mask Respect soft masking in the reference FASTA
-h, --help Print help
motif search
Search for modification-enriched subsequences in a reference genome
Usage: modkit motif search [OPTIONS] --in-bedmethyl <IN_BEDMETHYL> --ref <REFERENCE_FASTA>
Options:
--force-override-spec
Force override SAM specification of association of modification codes
to primary sequence bases
-h, --help
Print help (see a summary with '-h')
Input Options:
-i, --in-bedmethyl <IN_BEDMETHYL>
Input bedmethyl table, can be used directly from modkit pileup
-r, --ref <REFERENCE_FASTA>
Reference sequence in FASTA format used for the pileup
--contig <CONTIG>
Use only bedMethyl records from this contig, requires that the
bedMethyl be BGZIP-compressed and tabix-indexed
Compute Options:
-t, --threads <THREADS>
Number of threads to use
[default: 4]
--io-threads <IO_THREADS>
Number of tabix/bgzf IO threads to use
[default: 2]
Logging Options:
--log-filepath <LOG_FILEPATH>
Output log to this file
--suppress-progress
Disable the progress bars
Search Options:
--low-thresh <LOW_THRESHOLD>
Fraction modified threshold below which consider a genome location to
be "low modification"
[default: 0.2]
--high-thresh <HIGH_THRESHOLD>
Fraction modified threshold above which consider a genome location to
be "high modification" or enriched for modification
[default: 0.6]
--min-frac-mod <FRAC_SITES_THRESH>
Minimum fraction of sites in the genome to be "high-modification" for
a motif to be considered
[default: 0.85]
--context-size <CONTEXT_SIZE> <CONTEXT_SIZE>
Upstream and downstream number of bases to search for a motif sequence
around a modified base. Example: --context-size 12 12
[default: 12 12]
--min-coverage <MIN_COVERAGE>
Minimum valid coverage in the bedMethyl to consider a record valid
[default: 5]
--min-sites <MIN_SITES>
Minimum number of total sites in the genome required for a motif to be
considered
[default: 300]
--min-log-odds <MIN_LOG_ODDS>
Minimum log-odds to consider a motif sequence to be enriched
[default: 1.5]
--init-context-size <INIT_CONTEXT_SIZE> <INIT_CONTEXT_SIZE>
Initial "fixed" seed window size in base pairs around the modified
base. Example: --init-context-size 2 2
[default: 2 2]
--mod-code <MOD_CODES>
Specify which modification codes to process, default will process all
modification codes found in the input bedMethyl file
Output Options:
-o, --out-table <OUT_TABLE>
Optionally output a machine-parsable TSV (human-readable table will
always be output to the log)
--known-motif <KNOWN_MOTIFS> <KNOWN_MOTIFS> <KNOWN_MOTIFS>
Include statistics on a suspected or known motif. Format should be
<sequence> <offset> <mod_code>
--known-motifs-table <KNOWN_MOTIFS_TABLE>
Path to known motifs in tabular format. Tab-separated values:
<mod_code>\t<motif_seq>\t<offset>. May have the same header as the
output table from this command
--eval-motifs-table <OUT_KNOWN_TABLE>
Optionally output machine parsable table with known motif modification
frequencies that were not found during search
Exhaustive Search Options:
--exhaustive-seed-min-log-odds <EXHAUSTIVE_SEED_MIN_LOG_ODDS>
Minimum log-odds to consider a motif seed sequence to be enriched when
performing exhaustive search, decreasing this number will increase the
number of seeds searched and thus computational time
[default: 2.5]
--exhaustive-seed-len <EXHAUSTIVE_SEED_LEN>
Exhaustive search seed length, increasing this value increases
computational time
[default: 3]
--skip-search
Skip the exhaustive search phase, saves time but the results may be
less sensitive
--search-top-pct <SEARCH_TOP_PCT>
During exhaustive search, instead of searching all seeds with log-odds
above `exhaustive_seed_min_log_odds`, only search the top X-percent of
seeds. Can be used with `min_exhaustive_seeds` and
`max_exhaustive_seeds`
--narrow-search
When used in conjunction with `search_top_pct`, search the top
X-percent of seeds, and then narrow the search space by removing
contexts matching any motifs found. Then iterate until zero additional
motifs are found or another stopping condition is reached
--search-timeout <SEARCH_TIMEOUT>
A stopping condition when using `--narrow-search`, stop once exaustive
search for a modification code has been worked on for this long
--search-batch-size <SEARCH_BATCH_SIZE>
Set the batch size when performing a simple timeout on search. At
least this many seeds will be evaluated
[default: 100]
--max-exhaustive-seeds <MAX_EXHAUSTIVE_SEEDS>
Set the maximum number of exhaustive seeds to be searched in a batch.
Overrides the X-percent of seeds to be searched when that number
exceeds this setting
--min-exhaustive-seeds <MIN_EXHAUSTIVE_SEEDS>
Search at least this many seeds. Overrides the X-percent of seeds to
be searched when that number is less than this setting
[default: 20]
--max-narrow-iters <MAX_NARROW_ITERS>
Stopping condition when using `--narrow-search` and
`--search-top-pct`, stop after this many iterations regardless if the
timeout is provided and has been reached. Exaustive search will still
stop when once no more motifs are found
motif evaluate
Calculate enrichment statistics on a set of motifs from a bedMethyl table
Usage: modkit motif evaluate [OPTIONS] --in-bedmethyl <IN_BEDMETHYL> --ref <REFERENCE_FASTA>
Options:
--force-override-spec
Force override SAM specification of association of modification codes
to primary sequence bases
--suppress-table
Don't print final table to stderr (will still go to log file)
-h, --help
Print help (see a summary with '-h')
Input Options:
-i, --in-bedmethyl <IN_BEDMETHYL>
Input bedmethyl table, can be used directly from modkit pileup
-r, --ref <REFERENCE_FASTA>
Reference sequence in FASTA format used for the pileup
--contig <CONTIG>
Use only bedMethyl records from this contig, requires that the
bedMethyl be BGZIP-compressed and tabix-indexed
Compute Options:
-t, --threads <THREADS>
Number of threads to use
[default: 4]
--io-threads <IO_THREADS>
Number of tabix/bgzf IO threads to use
[default: 2]
Logging Options:
--log-filepath <LOG_FILEPATH>
Output log to this file
--suppress-progress
Disable the progress bars
Output Options:
--known-motif <KNOWN_MOTIFS> <KNOWN_MOTIFS> <KNOWN_MOTIFS>
Format should be <sequence> <offset> <mod_code>
--known-motifs-table <KNOWN_MOTIFS_TABLE>
Path to known motifs in tabular format. Tab-separated values:
<mod_code>\t<motif_seq>\t<offset>. May have the same header as the
output table from this command
--out <OUT_TABLE>
Machine-parsable table of refined motifs. Human-readable table always
printed to stderr and log
Search Options:
--min-coverage <MIN_COVERAGE>
Minimum valid coverage in the bedMethyl to consider a record valid
[default: 5]
--context-size <CONTEXT_SIZE> <CONTEXT_SIZE>
Upstream and downstream number of bases to search for a motif sequence
around a modified base. Example: --context-size 12 12
[default: 12 12]
--low-thresh <LOW_THRESHOLD>
Fraction modified threshold below which consider a genome location to
be "low modification"
[default: 0.2]
--high-thresh <HIGH_THRESHOLD>
Fraction modified threshold above which consider a genome location to
be "high modification" or enriched for modification
[default: 0.6]
motif refine
Use a previously defined list of motif sequences and further refine them with a
bedMethyl table
Usage: modkit motif refine [OPTIONS] --in-bedmethyl <IN_BEDMETHYL> --ref <REFERENCE_FASTA>
Options:
--min-refine-frac-mod <MIN_REFINE_FRAC_MODIFIED>
Minimum fraction of sites in the genome to be "high-modification" for
a motif to be further refined, otherwise it will be discarded
[default: 0.6]
--min-refine-sites <MIN_REFINE_SITES>
Minimum number of total sites in the genome required for a motif to be
further refined, otherwise it will be discarded
[default: 300]
--force-override-spec
Force override SAM specification of association of modification codes
to primary sequence bases
-h, --help
Print help (see a summary with '-h')
Input Options:
-i, --in-bedmethyl <IN_BEDMETHYL>
Input bedmethyl table, can be used directly from modkit pileup
-r, --ref <REFERENCE_FASTA>
Reference sequence in FASTA format used for the pileup
--contig <CONTIG>
Use only bedMethyl records from this contig, requires that the
bedMethyl be BGZIP-compressed and tabix-indexed
Compute Options:
-t, --threads <THREADS>
Number of threads to use
[default: 4]
--io-threads <IO_THREADS>
Number of tabix/bgzf IO threads to use
[default: 2]
Logging Options:
--log-filepath <LOG_FILEPATH>
Output log to this file
--suppress-progress
Disable the progress bars
Output Options:
--known-motif <KNOWN_MOTIFS> <KNOWN_MOTIFS> <KNOWN_MOTIFS>
Format should be <sequence> <offset> <mod_code>
--known-motifs-table <KNOWN_MOTIFS_TABLE>
Path to known motifs in tabular format. Tab-separated values:
<mod_code>\t<motif_seq>\t<offset>. May have the same header as the
output table from this command
--out <OUT_TABLE>
Machine-parsable table of refined motifs. Human-readable table always
printed to stderr and log
Search Options:
--low-thresh <LOW_THRESHOLD>
Fraction modified threshold below which consider a genome location to
be "low modification"
[default: 0.2]
--high-thresh <HIGH_THRESHOLD>
Fraction modified threshold above which consider a genome location to
be "high modification" or enriched for modification
[default: 0.6]
--min-frac-mod <FRAC_SITES_THRESH>
Minimum fraction of sites in the genome to be "high-modification" for
a motif to be considered
[default: 0.85]
--context-size <CONTEXT_SIZE> <CONTEXT_SIZE>
Upstream and downstream number of bases to search for a motif sequence
around a modified base. Example: --context-size 12 12
[default: 12 12]
--min-coverage <MIN_COVERAGE>
Minimum valid coverage in the bedMethyl to consider a record valid
[default: 5]
--min-sites <MIN_SITES>
Minimum number of total sites in the genome required for a motif to be
considered
[default: 300]
--min-log-odds <MIN_LOG_ODDS>
Minimum log-odds to consider a motif sequence to be enriched
[default: 1.5]
dmr pair
Compare regions in a pair of samples (for example, tumor and normal or control
and experiment). A sample is input as a bgzip pileup bedMethyl (produced by
pileup, for example) that has an associated tabix index. Output is a BED file
with the score column indicating the magnitude of the difference in methylation
between the two samples. See the online documentation for additional details
Usage: modkit dmr pair [OPTIONS] --ref <REFERENCE_FASTA>
Options:
-r, --regions-bed <REGIONS_BED>
BED file of regions over which to compare methylation levels. Should
be tab-separated (spaces allowed in the "name" column). Requires
chrom, chromStart and chromEnd. The Name column is optional. Strand is
currently ignored. When omitted, methylation levels are compared at
each site
--ref <REFERENCE_FASTA>
Path to reference fasta for used in the pileup/alignment
-h, --help
Print help (see a summary with '-h')
Sample Options:
-a <CONTROL_BED_METHYL>
Bgzipped bedMethyl file for the first (usually control) sample. There
should be a tabix index with the same name and .tbi next to this file
or the --index-a option must be provided
-b <EXP_BED_METHYL>
Bgzipped bedMethyl file for the second (usually experimental) sample.
There should be a tabix index with the same name and .tbi next to this
file or the --index-b option must be provided
-m, --base <MODIFIED_BASES>
Bases to use to calculate DMR, may be multiple. For example, to
calculate differentially methylated regions using only cytosine
modifications use --base C
--assign-code <MOD_CODE_ASSIGNMENTS>
Extra assignments of modification codes to their respective primary
bases. In general, modkit dmr will use the SAM specification to know
which modification codes are appropriate to use for a given primary
base. For example "h" is the code for 5hmC, so is appropriate for
cytosine bases, but not adenine bases. However, if your bedMethyl file
contains custom codes or codes that are not part of the specification,
you can specify which primary base they belong to here with
--assign-code x:C meaning associate modification code "x" with
cytosine (C) primary sequence bases. If a code is encountered that is
not part of the specification, the bedMethyl record will not be used,
this will be logged
--single-code <SINGLE_MODIFICATION_CODE>
Calculate differential methylation for this modification code in
isolation
-k, --mask
Respect soft masking in the reference FASTA
--min-valid-coverage <MIN_VALID_COVERAGE>
Minimum valid coverage required to use an entry from a bedMethyl. See
the help for pileup for the specification and description of valid
coverage
[default: 0]
Output Options:
-o, --out-path <OUT_PATH>
Path to file to direct output, optional, no argument will direct
output to stdout
--header
Include header in output
Segmentation Options:
--segment <SEGMENTATION_FP>
Run segmentation, output segmented differentially methylated regions
to this file
--max-gap-size <MAX_GAP_SIZE>
Maximum number of base pairs between modified bases for them to be
segmented together
[default: 5000]
--dmr-prior <DMR_PRIOR>
Prior probability of a differentially methylated position
[default: 0.1]
--diff-stay <DIFF_STAY>
Maximum probability of continuing a differentially methylated block,
decay will be dynamic based on proximity to the next position
[default: 0.9]
--significance-factor <SIGNIFICANCE_FACTOR>
Significance factor, effective p-value necessary to favor the
"Different" state
[default: 0.01]
--log-transition-decay
Use logarithmic decay for "Different" stay probability
--decay-distance <DECAY_DISTANCE>
After this many base pairs, the transition probability will become the
prior probability of encountering a differentially modified position
[default: 500]
--fine-grained
Preset HMM segmentation parameters for higher propensity to switch
from "Same" to "Different" state. Results will be shorter segments,
but potentially higher sensitivity
Logging Options:
--careful
Log out which sequences are in common between the samples and the
reference FASTA, useful for debugging
--log-filepath <LOG_FILEPATH>
File to write logs to, it's recommended to use this option
--suppress-progress
Don't show progress bars
--missing <HANDLE_MISSING>
How to handle regions found in the `--regions` BED file. quiet =>
ignore regions that are not found in the tabix header warn => log
(debug) regions that are missing fatal => log (error) and exit the
program when a region is missing
[default: quiet]
[possible values: quiet, warn, fail]
Compute Options:
-t, --threads <THREADS>
Number of threads to use
[default: 4]
--io-threads <IO_THREADS>
Number of threads to use when for decompression
[default: 4]
--batch-size <BATCH_SIZE>
Control the batch size. The batch size is the number of regions to
load at a time. Each region will be processed concurrently. Loading
more regions at a time will decrease IO to load data, but will use
more memory. Default will be 50% more than the number of threads
assigned
-f, --force
Force overwrite of output file, if it already exists
-i, --interval-size <INTERVAL_SIZE>
Interval chunk size in base pairs to process concurrently. Smaller
interval chunk sizes will use less memory but incur more overhead
[default: 100000]
Single-site Options:
--prior <PRIOR> <PRIOR>
Prior distribution for estimating MAP-based p-value. Should be two
arguments for alpha and beta (e.g. 1.0 1.0). See
`dmr_scoring_details.md` for additional details on how the metric is
calculated
--delta <DELTA>
Consider only effect sizes greater than this when calculating the
MAP-based p-value
[default: 0.05]
-N, --n-sample-records <N_SAMPLE_RECORDS>
Sample this many reads when estimating the max coverage thresholds
[default: 10042]
--max-coverages <MAX_COVERAGES> <MAX_COVERAGES>
Max coverages to enforce when calculating estimated MAP-based p-value
--cap-coverages
When using replicates, cap coverage to be equal to the maximum
coverage for a single sample. For example, if there are 3 replicates
with max_coverage of 30, the total coverage would normally be 90.
Using --cap-coverages will down sample the data to 30X
dmr multi
Compare regions between all pairs of samples (for example a trio sample set or
haplotyped trio sample set). As with `pair` all inputs must be bgzip compressed
bedMethyl files with associated tabix indices. Each sample must be assigned a
name. Output is a directory of BED files with the score column indicating the
magnitude of the difference in methylation between the two samples indicated in
the file name. See the online documentation for additional details
Usage: modkit dmr multi [OPTIONS] --regions-bed <REGIONS_BED> --out-dir <OUT_DIR> --ref <REFERENCE_FASTA>
Options:
-h, --help Print help
Sample Options:
-s, --sample <SAMPLES> <SAMPLES>
Two or more named samples to compare. Two arguments are required
<path> <name>. This option should be repeated at least two times. When
two samples have the same name, they will be combined
-r, --regions-bed <REGIONS_BED>
BED file of regions over which to compare methylation levels. Should
be tab-separated (spaces allowed in the "name" column). Requires
chrom, chromStart and chromEnd. The Name column is optional. Strand is
currently ignored
--ref <REFERENCE_FASTA>
Path to reference fasta for the pileup
-m, --base <MODIFIED_BASES>
Bases to use to calculate DMR, may be multiple. For example, to
calculate differentially methylated regions using only cytosine
modifications use --base C
--assign-code <MOD_CODE_ASSIGNMENTS>
Extra assignments of modification codes to their respective primary
bases. In general, modkit dmr will use the SAM specification to know
which modification codes are appropriate to use for a given primary
base. For example "h" is the code for 5hmC, so is appropriate for
cytosine bases, but not adenine bases. However, if your bedMethyl file
contains custom codes or codes that are not part of the specification,
you can specify which primary base they belong to here with
--assign-code x:C meaning associate modification code "x" with
cytosine (C) primary sequence bases. If a code is encountered that is
not part of the specification, the bedMethyl record will not be used,
this will be logged
--single-code <SINGLE_MODIFICATION_CODE>
Calculate differential methylation for this modification code in
isolation
-k, --mask
Respect soft masking in the reference FASTA
--min-valid-coverage <MIN_VALID_COVERAGE>
Minimum valid coverage required to use an entry from a bedMethyl. See
the help for pileup for the specification and description of valid
coverage [default: 0]
--ref-sample <REF_SAMPLE>
Only performs comparisons between this sample and all other samples
Output Options:
--header Include header in output
-o, --out-dir <OUT_DIR> Directory to place output DMR results in BED format
-p, --prefix <PREFIX> Prefix files in directory with this label
-f, --force Force overwrite of output file, if it already exists
Logging Options:
--log-filepath <LOG_FILEPATH>
File to write logs to, it's recommended to use this option
--suppress-progress
Don't show progress bars
--missing <HANDLE_MISSING>
How to handle regions found in the `--regions` BED file. quiet =>
ignore regions that are not found in the tabix header warn => log
(debug) regions that are missing fatal => log (error) and exit the
program when a region is missing [default: quiet] [possible values:
quiet, warn, fail]
Compute Options:
-t, --threads <THREADS>
Number of threads to use [default: 4]
--io-threads <IO_THREADS>
Number of threads to use when for decompression [default: 4]
dmr isoform
Use a transcriptome-aligned bedMethyl table and GTF file to compare methylation
across gene transcript isoforms. Run analysis on the entire transcriptome or on
a single gene. Optionally also make plots for single genes
Usage: modkit dmr isoform [OPTIONS] --gtf <GTF> <INPUT_BEDMETHYL> <OUTPUT_TABLE>
Arguments:
<INPUT_BEDMETHYL>
Input bedmethyl table. Expected to be bgzip-compressed and have a .tbi
tabix index. This bedMethyl table must be generated from a
transcriptome-aligned modBAM, NOT a genome-aligned modBAM. Expected
that this table was generated by pileup with `--modified-bases`
argument
<OUTPUT_TABLE>
Path to output BED file or '-' for standard output
Options:
--gtf <GTF>
Path to GTF file to use for transcript models. May be compressed
--min-valid-coverage <MIN_VALID_COVERAGE>
Minimum valid coverage required to use an entry from a bedMethyl. See
the help for pileup for the specification and description of valid
coverage
[default: 1]
--single-code <SINGLE_MODIFICATION_CODE>
Calculate differential methylation for this modification code in
isolation. Unmodified and other modification counts will be combined
--full
Include per-modification combined proportions per position and
per-isoform proportions in output. Adding this flag will make the
output file substantially larger
--ignore-version
Ignore gene ID version and transcript ID version in GTF, if there are
multiple identical transcript IDs or gene IDs with different versions
the last one will be kept
--log-filepath <LOG_FILEPATH>
Path to optional debug log (recommended)
-g, --gene-id <GENE_ID>
Only process this Gene Id
-n, --gene-name <GENE_NAME>
Only process this Gene Name
--gene-strand <GENE_STRAND>
Specify a strand for the Gene Name (or GeneID) if there are multiple
chromosomes with this gene
--gene-chrom <GENE_CHROM>
Specify a chromosome for the Gene Name (or GeneID) if there are
multiple chromosomes with this gene
--suppress-progress
Don't show the progress bar
-t, --threads <THREADS>
Number of concurrent processors to use. Only used when running on all
genes
[default: 4]
--plot <PLOT>
Generate an SVG plot displaying the methylation positions on each
isoform (filtered by --min-score or --max-pval) and the common exons
of the gene. Also displays negative log of the p-value of the DMR
metric. See the online documentation for an example
--marker-positions <MARKER_POSITIONS>...
Comma-separated list of genomic positions to draw a vertical dotted
line and a label
--width <WIDTH>
SVG width in px
[default: 1600]
--track-height <TRACK_HEIGHT>
Height per transcript track in px
[default: 26]
--track-gap <TRACK_GAP>
Gap between transcript tracks in px
[default: 18]
--left-margin <LEFT_MARGIN>
Left margin in px
[default: 220]
--right-margin <RIGHT_MARGIN>
Right margin in px
[default: 40]
--top-margin <TOP_MARGIN>
Top margin in px
[default: 210]
--bottom-margin <BOTTOM_MARGIN>
Bottom margin in px
[default: 50]
--intron-px <INTRON_PX>
Length, in px, of the compressed intron segments
[default: 24]
--top-bar-height <TOP_BAR_HEIGHT>
Height in px of the top bar
[default: 90]
--min-score <MIN_SCORE>
Only plot methylation markers with negative log p-value greater than
this value
--max-pval <MAX_PVAL>
Only plot methylation markers with p-value less than this value
-h, --help
Print help (see a summary with '-h')
dmr compare-tx-sites
Uses two transcriptome-aligned bedmMethyl tables to compare methylation for two
conditions at transcriptome sites aggregated at the gene level. This command
will combine the methylation counts across isoforms into a single summary
statistic per-site for the gene, then compare those counts between conditions at
the gene level and report results on genomic coordinates. The transcript models
are provided by a GTF file. Optionally make a volcano plot of the top-k
differentially methylated sites per gene
Usage: modkit dmr compare-tx-sites [OPTIONS] -a <COND_A> -b <COND_B> --out <OUTPUT_TABLE> --gtf <GTF>
Options:
-a <COND_A>
Input bedmethyl table for first condition (condition 'A'). Expected to
be bgzip-compressed and have a .tbi tabix index. This bedMethyl table
must be generated from a transcriptome-aligned modBAM, NOT a
genome-aligned modBAM. Expected that this table was generated by
pileup with `--modified-bases` argument
-b <COND_B>
Input bedmethyl table for first condition (condition 'B')
-o, --out <OUTPUT_TABLE>
Path to output BED file or '-' for stdout
--full
Include combined proportions and counts for each condition. This will
increase the size of the output file
--gtf <GTF>
Path to GTF file to use for transcript models. Can be compressed
--ignore-version
Ignore gene ID version and transcript ID version in GTF, if there are
multiple identical transcript IDs or gene IDs with different versions
the last one will be kept
--min-valid-coverage <MIN_VALID_COVERAGE>
Minimum valid coverage required to use an entry from a bedMethyl. See
the help for pileup for the specification and description of valid
coverage [default: 1]
--single-code <SINGLE_MODIFICATION_CODE>
Calculate differential methylation for this modification code in
isolation. Unmodified and other modification counts will be combined
--log-filepath <LOG_FILEPATH>
Path to optional debug log (recommended)
--suppress-progress
Don't show the progress bar
-t, --threads <THREADS>
Number of concurrent processors to use [default: 4]
--plot <PLOT>
Path to file to create an SVG volcano plot where the x-axis is log2
fold change and the y-axis is negative log p-value. See the online
documentation for and example
--top-k <TOP_K>
For each gene, sort the positions in increasing p-value and take this
many for plotting. Setting a large number will increase the size of
the SVG plot [default: 1]
--label-top-k-genes <LABEL_TOP_K_GENES>
Sort the points to be plotted by increasing p-value, then label the
points from the top-k genes
--min-effect-size <MIN_EFFECT_SIZE>
Discard positions with an effect size less than this before
considering them for plotting [default: 0.1]
--plot-title <PLOT_TITLE>
Add a title to the plot
--gene-labels <GENE_LABELS>
Path to plain text file of a list of gene names, one per line, to mark
in the volcano plot
--sort-by-effect-size
Determine which points to plot by sorting to decreasing effect size
instead of increasing p-value
-h, --help
Print help
bedmethyl merge
Perform an outer join on two or more bedMethyl files, summing their counts for
records that overlap
Usage: modkit bedmethyl merge [OPTIONS] --out-bed <OUT_BED> --genome-sizes <GENOME_SIZES> [IN_BEDMETHYL] [IN_BEDMETHYL]...
Arguments:
[IN_BEDMETHYL] [IN_BEDMETHYL]...
Input bedMethyl table(s). Should be bgzip-compressed and have an
associated Tabix index. The tabix index will be assumed to be
$this_file.tbi
Options:
-g, --genome-sizes <GENOME_SIZES>
TSV of genome sizes, should be <chrom>\t<size_in_bp>
-h, --help
Print help (see a summary with '-h')
Output Options:
-o, --out-bed <OUT_BED>
Specify the output file to write the results table
--force
Force overwrite the output file
--header
Output a header with the bedMethyl
--mixed-delim
Output bedMethyl where the delimiter of columns past column 10 are
space-delimited instead of tab-delimited. This option can be useful
for some browsers and parsers that don't expect the extra columns of
the bedMethyl format
Compute Options:
--chunk-size <CHUNK_SIZE>
Chunk size for how many start..end regions for each chromosome to
read. Larger values will lead to faster merging at the expense of
memory usage, while smaller values will be slower with lower memory
usage. This option will only impact large bedmethyl files
-i, --interval-size <INTERVAL_SIZE>
Interval chunk size in base pairs to process concurrently. Smaller
interval chunk sizes will use less memory but incur more overhead
[default: 100000]
-t, --threads <THREADS>
Number of threads to use
[default: 4]
--queue-size <QUEUE_SIZE>
Number of batches (of size chunk size) allowed to be in a pre-written
state at once. Increasing this number will increase memory usage
[default: 30]
--io-threads <IO_THREADS>
Number of tabix/bgzf threads to use
[default: 2]
Logging Options:
--log-filepath <LOG_FILEPATH>
Specify a file to write debug logs to
Filtering Options:
--min-samples <MIN_SAMPLES>
Only output a position present in at least this many input bedMethyl
files. Accepts an integer greater than 1, or "all" to require the
position in every input (an inner join across replicates)
--min-sample-coverage <MIN_SAMPLE_COVERAGE>
Minimum valid coverage for an input's record to count towards a
position, for both the --min-samples tally and the summed counts
bedmethyl tobigwig
Make a BigWig track from a bedMethyl file or stream. For details on the BigWig
format see https://doi.org/10.1093/bioinformatics/btq351
Usage: modkit bedmethyl tobigwig [OPTIONS] --mod-codes <MOD_CODES> <--sizes <CHROMSIZES>|--header <INPUT_BAM>> <IN_BEDMETHYL> <OUT_FP>
Arguments:
<IN_BEDMETHYL>
Input bedmethyl, uncompressed, "-" or "stdin" indicates an input
stream
<OUT_FP>
Output bigWig filename
Options:
-g, --sizes <CHROMSIZES>
A chromosome sizes file. Each line should be a chromosome and its size
in bases, separated by whitespace. A fasta index (.fai) works as well.
Use instead of the bam header
-b, --header <INPUT_BAM>
modBAM from which the pileup was generated. Chromosome sizes are
gathered from the header. Use instead of the chromosome sizes file
-m, --mod-codes <MOD_CODES>
Make a bigWig track where the values are the percent of bases with
this modification, use multiple comma-separated codes to combine
counts. For example --mod-code m makes a track of the 5mC percentages
and --mod-codes h,m will make a track of the combined counts from 5hmC
and 5mC. Combining counts for different primary bases will cause an
error (e.g. --mod-codes a,h will error)
-h, --help
Print help (see a summary with '-h')
Output Options:
--negative-strand-values
Report the percentages on the negative strand as negative values. The
data range will be [-100, 100]
-z, --nzooms <NZOOMS>
Set the maximum of zooms to create
[default: 10]
--zooms <ZOOMS>...
Set the zoom resolutions to use (overrides the --nzooms argument)
-u, --uncompressed
Don't use compression
--block-size <BLOCK_SIZE>
Number of items to bundle in r-tree
[default: 256]
--items-per-slot <ITEMS_PER_SLOT>
Number of data points bundled at lowest level
[default: 1024]
Compute Options:
-t, --nthreads <NTHREADS>
Set the number of threads to use. This tool will typically use ~225%
CPU on a HDD. SDDs may be higher. (IO bound)
[default: 6]
--inmemory
Do not create temporary files for intermediate data
--force-chromosome-ordering
If input bedMethyl has sorting of the same scheme as `sort`, this
option may speed up conversion
Logging Options:
--log-filepath <LOG_FILEPATH>
Specify a file for debug logs to be written to, otherwise ignore them.
Setting a file is recommended. (alias: log)
--suppress-progress
Hide the progress bar
bedmethyl map-to-genome
Map a transcriptome-aligned bedMethyl file to genome-coordinates based on a GTF
Usage: modkit bedmethyl map-to-genome [OPTIONS] --transcript-id <TRANSCRIPT_ID> --gtf <GTF> <INPUT_BEDMETHYL> <OUTPUT_BEDMETHYL>
Arguments:
<INPUT_BEDMETHYL>
<OUTPUT_BEDMETHYL>
Options:
-t, --transcript-id <TRANSCRIPT_ID>
--transcript-version <TRANSCRIPT_VERSION>
--ignore-version
Ignore gene ID version and transcript ID version in GTF, if there are
multiple identical transcript IDs or gene IDs with different versions
the last one will be kept
--gtf <GTF>
--header
-h, --help
Print help
modbam adjust-mods
Performs various operations on BAM files containing base modification
information, such as converting base modification codes and ignoring
modification calls. Produces a BAM output file
Usage: modkit modbam adjust-mods [OPTIONS] <IN_BAM> <OUT_BAM>
Arguments:
<IN_BAM>
Input BAM file, can be a path to a file or one of `-` or `stdin` to
specify a stream from standard input
<OUT_BAM>
File path to new BAM file to be created. Can be a path to a file or
one of `-` or `stdin` to specify a stream from standard output
Options:
-f, --ff
Fast fail, stop processing at the first invalid sequence record.
Default behavior is to continue and report failed/skipped records at
the end
-h, --help
Print help (see a summary with '-h')
Output Options:
--log-filepath <LOG_FILEPATH>
Output debug logs to file at this path
--output-sam
Output SAM format instead of BAM
Modified Base Options:
--ignore <IGNORE>
Modified base code to ignore/remove, see
https://samtools.github.io/hts-specs/SAMtags.pdf for details on the
modified base codes
--convert <CONVERT> <CONVERT>
Convert one mod-tag to another, summing the probabilities together if
the retained mod tag is already present
--motif <MOTIF> <MOTIF>
Filter out any base modification call that isn't part of a basecall
sequence motif. This argument can be passed multiple times. Format is
<motif_sequence> <offset>. For example the argument to match CpG
dinucleotides is `--motif CG 0`, or to match CG[5mC]G the argument
would be `--motif CGCG 2`. Single bases can be used as motifs to keep
only base modification calls for a specific primary base, for example
`--motif C 0`
--cpg
Shorthand for --motif CG 0
--discard-motifs
Discard base modification calls that match the provided motifs
(instead of keeping them)
Compute Options:
-t, --threads <THREADS>
Number of threads to use
[default: 4]
Selection Options:
--edge-filter <EDGE_FILTER>
Discard base modification calls that are this many bases from the
start or the end of the read. Two comma-separated values may be
provided to asymmetrically filter out base modification calls from the
start and end of the reads. For example, 4,8 will filter out base
modification calls in the first 4 and last 8 bases of the read
--invert-edge-filter
Invert the edge filter, instead of filtering out base modification
calls at the ends of reads, only _keep_ base modification calls at the
ends of reads. E.g. if usually, "4,8" would remove (i.e. filter out)
base modification calls in the first 4 and last 8 bases of the read,
using this flag will keep only base modification calls in the first 4
and last 8 bases
--filter-probs
Filter out the lowest confidence base modification probabilities
--mapped-only
Only use base modification probabilities from bases that are aligned
when estimating the filter threshold (i.e. ignore soft-clipped, and
inserted bases)
Sampling Options:
-n, --num-reads <NUM_READS>
Sample approximately this many reads when estimating the filtering
threshold. If alignments are present reads will be sampled evenly
across aligned genome. If a region is specified, either with the
--region option or the --sample-region option, then reads will be
sampled evenly across the region given. This option is useful for
large BAM files. In practice, 10-50 thousand reads is sufficient to
estimate the model output distribution and determine the filtering
threshold
[default: 10042]
--sample-region <SAMPLE_REGION>
Specify a region for sampling reads from when estimating the threshold
probability. If this option is not provided, but --region is provided,
the genomic interval passed to --region will be used. Format should be
<chrom_name>:<start>-<end> or <chrom_name>
--sampling-interval-size <SAMPLING_INTERVAL_SIZE>
Interval chunk size to process concurrently when estimating the
threshold probability, can be larger than the pileup processing
interval
[default: 1000000]
Filtering Options:
-p, --filter-percentile <FILTER_PERCENTILE>
Filter out modified base calls where the probability of the predicted
variant is below this confidence percentile. For example, 0.1 will
filter out the 10% lowest confidence modification calls
[default: 0.1]
--filter-threshold <FILTER_THRESHOLD>
Specify the filter threshold globally or per primary base. A global
filter threshold can be specified with by a decimal number (e.g.
0.75). Per-base thresholds can be specified by colon-separated values,
for example C:0.75 specifies a threshold value of 0.75 for cytosine
modification calls. Additional per-base thresholds can be specified by
repeating the option: for example --filter-threshold C:0.75
--filter-threshold A:0.70 or specify a single base option and a
default for all other bases with: --filter-threshold A:0.70
--filter-threshold 0.9 will specify a threshold value of 0.70 for
adenine and 0.9 for all other base modification calls
--mod-threshold <MOD_THRESHOLDS>
Specify a passing threshold to use for a base modification,
independent of the threshold for the primary sequence base or the
default. For example, to set the pass threshold for 5hmC to 0.8 use
`--mod-threshold h:0.8`. The pass threshold will still be estimated as
usual and used for canonical cytosine and other modifications unless
the `--filter-threshold` option is also passed. See the online
documentation for more details
Logging Options:
--suppress-progress
Hide the progress bar
modbam update-tags
Renames Mm/Ml to tags to MM/ML. Also allows changing the mode flag from silent
'.' to explicitly '?' or '.'
Usage: modkit modbam update-tags [OPTIONS] <IN_BAM> <OUT_BAM>
Arguments:
<IN_BAM> BAM to update modified base tags in. Can be a path to a file or one
of `-` or `stdin` to specify a stream from standard input
<OUT_BAM> File to new BAM file to be created or one of `-` or `stdin` to
specify a stream from standard output
Options:
-m, --mode <MODE> Mode, change mode to this value, options {'explicit',
'implicit'}. See spec at:
https://samtools.github.io/hts-specs/SAMtags.pdf.
'explicit' ('?') means residues without modification
probabilities will not be assumed canonical or
modified. 'implicit' means residues without explicit
modification probabilities are assumed to be
canonical [possible values: explicit, implicit]
--no-implicit-probs Don't add implicit canonical calls. This flag is
important when converting from one of the implicit
modes ( `.` or `""`) to explicit mode (`?`). By
passing this flag, the bases without associated base
modification probabilities will not be assumed to be
canonical. No base modification probability will be
written for these bases, meaning there is no
information. The mode will automatically be set to
the explicit mode `?`
-h, --help Print help
Compute Options:
-t, --threads <THREADS> Number of threads to use [default: 4]
Logging Options:
--log-filepath <LOG_FILEPATH> Output debug logs to file at this path
Output Options:
--output-sam Output SAM format instead of BAM
modbam sample-probs
Calculate an estimate of the base modification probability distribution
Usage: modkit modbam sample-probs [OPTIONS] <IN_BAM>
Arguments:
<IN_BAM>
Input BAM with modified base tags. If a index is found reads will be
sampled evenly across the length of the reference sequence. Can be a
path to a file or one of `-` or `stdin` to specify a stream from
standard input
Options:
--ignore-index
Ignore the BAM index (if it exists) perform a serial scan of the BAM,
otherwise if an index is found, multiple workers will be used to read
the BAM in parallel. Conflicts with --threads since there will only be
one reader
-r, --reference <REFERENCE_FASTA>
Reference sequence in FASTA format. (alias: 'ref')
--preload-references
Preload the reference sequences, useful when working with many, short
reference sequences such as a transcriptome
-k, --mask
Respect soft masking in the reference FASTA
-h, --help
Print help (see a summary with '-h')
Compute Options:
-t, --threads <THREADS>
Number of workers to run concurrently. When an aligned, indexed BAM is
found, multiple workers (threads) will be spawned to read disjoint
sections of the BAM in parallel
[default: 4]
--io-threads <IO_THREADS>
Number of IO/decompression threads to use, these are shared across
workers (if an indexed BAM is found) or used for a single reader if
--ignore-index is passed, stdin is used, or an index is not found
[default: 4]
-i, --interval-size <INTERVAL_SIZE>
Interval chunk size in base pairs to process concurrently. Smaller
interval chunk sizes will use less memory but incur more overhead.
Only used when sampling probs from an indexed bam
[default: 1000000]
Logging Options:
--log-filepath <LOG_FILEPATH>
Specify a file for debug logs to be written to, otherwise ignore them.
Setting a file is recommended
--suppress-progress
Hide the progress bar
Output Options:
-p, --percentiles <PERCENTILES>
Percentiles to calculate, a space separated list of floats (for
example: "10,20,90")
[default: 10,50,90]
-q, --quantiles <QUANTILES>
Quantiles to calculate, a space separated list of floats (for example:
"0.1,0.2,0.9")
-o, --out-dir <OUT_DIR>
Directory to deposit result tables into. Required for model
probability histogram output
--prefix <PREFIX>
Label to prefix output files with
--force
Overwrite results if present
--hist
Output histogram of base modification prediction probabilities
--dna-color <PRIMARY_BASE_COLORS> <PRIMARY_BASE_COLORS>
Set colors of primary bases in histogram, should be RGB format, e.g.
"#0000FF" is defailt for canonical cytosine
--mod-color <MOD_BASE_COLORS> <MOD_BASE_COLORS>
Set colors of modified bases in histogram, should be RGB format, e.g.
"#FF00FF" is default for 5hmC
Selection Options:
--motif <MOTIF> <MOTIF>
Tabulate base modification probabilities at motifs (or single bases).
The first argument should be the sequence motif and the second
argument is the 0-based offset to the base to pileup base modification
counts for. For example: --motif CGCG 0 indicates to collect base
mofification probabilities for the first C on the top strand and the
last C (complement to G) on the bottom strand. For single bases,
--motif C 0 or --motif A 0 would restrict to probabilities on C or A
reference bases, respectively. This argument can be passed multiple
times
--edge-filter <EDGE_FILTER>
Discard base modification calls that are this many bases from the
start or the end of the read. Two comma-separated values may be
provided to asymmetrically filter out base modification calls from the
start and end of the reads. For example, 4,8 will filter out base
modification calls in the first 4 and last 8 bases of the read
--region <REGION>
Process only the specified region of the BAM when collecting
probabilities. Format should be <chrom_name>:<start>-<end> or
<chrom_name>
--include-bed <INCLUDE_BED>
Only sample base modification probabilities that are aligned to the
positions in this BED file. (alias: include-positions)
--use-non-primary
Use non-primary mappings, may cause double-counting of reads with
primary and one or more non-primary mappings. Requires MN tag to be
correct. See documentation for help on proper alignment
Sampling Options:
-n, --num-reads <NUM_READS>
-f, --sampling-frac <SAMPLING_FRAC>
Instead of using a defined number of reads, specify a fraction of
reads to sample, for example 0.1 will sample 1/10th of the reads
--no-sampling
No sampling, use all of the reads to calculate the filter thresholds
-s, --seed <SEED>
Random seed for deterministic running, the default is
non-deterministic, only used when no BAM index is provided
modbam summary
Summarize the mod tags present in a BAM and get basic statistics. The default
output is a totals table (designated by '#' lines) and a modification calls
table. Descriptions of the columns can be found in the README
Usage: modkit modbam summary [OPTIONS] <IN_BAM>
Arguments:
<IN_BAM>
Input modBam, can be a path to a file or one of `-` or `stdin` to
specify a stream from standard input
Options:
--ignore-index
Ignore the BAM index (if it exists) perform a serial scan of the BAM,
otherwise if an index is found, multiple workers will be used to read
the BAM in parallel. Conflicts with --threads since there will only be
one reader
-r, --reference <REFERENCE_FASTA>
Reference sequence in FASTA format. (alias: 'ref')
--preload-references
Preload the reference sequences, useful when working with many, short
reference sequences such as a transcriptome
-k, --mask
Respect soft masking in the reference FASTA
--cpg
Shorthand for --motif CG 0. Requires a sorted, indexed modBAM
-h, --help
Print help (see a summary with '-h')
Compute Options:
-t, --threads <THREADS>
Number of threads to use
[default: 4]
--io-threads <IO_THREADS>
Number of IO/decompression threads to use, these are shared across
workers (if an indexed BAM is found) or used for a single reader if
--ignore-index is passed, stdin is used, or an index is not found
[default: 4]
-i, --interval-size <INTERVAL_SIZE>
When using regions, interval chunk size in base pairs to process
concurrently. Smaller interval chunk sizes will use less memory but
incur more overhead
[default: 1000000]
Logging Options:
--log-filepath <LOG_FILEPATH>
Specify a file for debug logs to be written to, otherwise ignore them.
Setting a file is recommended
--suppress-progress
Hide the progress bar
Output Options:
--tsv
Output summary as a tab-separated variables stdout instead of a table
--combine-mods
Combine modification counts per primary sequence base when calculating
summary statistics
Sampling Options:
-n, --num-reads <NUM_READS>
Approximate maximum number of reads to use. If an indexed BAM is
provided, the reads will be sampled evenly over the length of the
aligned reference. If a region is passed with the --region option,
they will be sampled over the region. Actual number of reads used may
deviate slightly from this number
-f, --sampling-frac <SAMPLING_FRAC>
Instead of using a defined number of reads, specify a fraction of
reads to sample when estimating the filter threshold. For example 0.1
will sample 1/10th of the reads
-s, --seed <SEED>
Sets a random seed for deterministic running (when using
--sample-frac)
Filtering Options:
-p, --filter-quantile <FILTER_QUANTILE>
Filter out modified base calls where the probability of the predicted
variant is below this confidence percentile. For example, 0.1 will
filter out the 10% lowest confidence base modification calls
[default: 0.1]
--filter-threshold <FILTER_THRESHOLD>
Specify the filter threshold globally or per-base. Global filter
threshold can be specified with by a decimal number (e.g. 0.75).
Per-base thresholds can be specified by colon-separated values, for
example C:0.75 specifies a threshold value of 0.75 for cytosine
modification calls. Additional per-base thresholds can be specified by
repeating the option: for example --filter-threshold C:0.75
--filter-threshold A:0.70 or specify a single base option and a
default for all other bases with: --filter-threshold A:0.70
--filter-threshold 0.9 will specify a threshold value of 0.70 for
adenine and 0.9 for all other base modification calls
--mod-threshold <MOD_THRESHOLDS>
Specify a passing threshold to use for a base modification,
independent of the threshold for the primary sequence base or the
default. For example, to set the pass threshold for 5hmC to 0.8 use
`--mod-threshold h:0.8`. The pass threshold will still be estimated as
usual and used for canonical cytosine and other modifications unless
the `--filter-threshold` option is also passed. See the online
documentation for more details
--max-filter-threshold <MAX_FILTER_THRESHOLD>
Maximum threhsold value to use, if the estimated threshold value for
any primary sequence base is greater than this value, use this value
instead. To set a default value for all primary bases, use
--max-threshold 0.8, for example. Or use per-base threshold values
like --max-threshold C:0.8
Selection Options:
--edge-filter <EDGE_FILTER>
Discard base modification calls that are this many bases from the
start or the end of the read. Two comma-separated values may be
provided to asymmetrically filter out base modification calls from the
start and end of the reads. For example, 4,8 will filter out base
modification calls in the first 4 and last 8 bases of the read
--include-bed <INCLUDE_BED>
Only summarize base modification probabilities that are aligned to the
positions in this BED file. (alias: include-positions)
--mapped-only
Only use modBAM records that are mapped, don't include unmapped
records
--matched-only
Only summarize base modifications that are mapped and are matches to
the appropriate primary sequence base. For example, require that 5mC
base modification calls be aligned to a reference C. Requires a
reference FASTA and an sorted, indexed modBAM
--motif <MOTIF> <MOTIF>
Calculate base modification statistics at reference motifs. This
option has the same behavior as "--matched-only" except it only
includes specified motifs. The first argument should be the sequence
motif and the second argument is the 0-based offset to the base to
pileup base modification counts for. For example: --motif CGCG 0
indicates to collect base mofification probabilities for the first C
on the top strand and the last C (complement to G) on the bottom
strand. For single bases, --motif C 0 or --motif A 0 would restrict to
probabilities on C or A reference bases, respectively. This argument
can be passed multiple times. Requires a sorted, indexed modBAM
--region <REGION>
Process only the specified region of the BAM when collecting
probabilities. Format should be <chrom_name>:<start>-<end> or
<chrom_name>
modbam call-mods
Call mods from a modbam, creates a new modbam with probabilities set to 100% if
a base modification is called or 0% if called canonical
Usage: modkit modbam call-mods [OPTIONS] <IN_BAM> <OUT_BAM>
Arguments:
<IN_BAM>
Input BAM, may be sorted and have associated index available. Can be a
path to a file or one of `-` or `stdin` to specify a stream from
standard input
<OUT_BAM>
Output BAM, can be a path to a file or one of `-` or `stdin` to
specify a stream from standard input
Options:
--log-filepath <LOG_FILEPATH>
Specify a file for debug logs to be written to, otherwise ignore them.
Setting a file is recommended
--ff
Fast fail, stop processing at the first invalid sequence record.
Default behavior is to continue and report failed/skipped records at
the end
--suppress-progress
Hide the progress bar
-t, --threads <THREADS>
Number of threads to use while processing chunks concurrently
[default: 4]
-n, --num-reads <NUM_READS>
Sample approximately this many reads when estimating the filtering
threshold. If alignments are present reads will be sampled evenly
across aligned genome. If a region is specified, either with the
--region option or the --sample-region option, then reads will be
sampled evenly across the region given. This option is useful for
large BAM files. In practice, 10-50 thousand reads is sufficient to
estimate the model output distribution and determine the filtering
threshold
[default: 10042]
-f, --sampling-frac <SAMPLING_FRAC>
Sample this fraction of the reads when estimating the
filter-percentile. In practice, 50-100 thousand reads is sufficient to
estimate the model output distribution and determine the filtering
threshold. See filtering.md for details on filtering
--seed <SEED>
Set a random seed for deterministic running, the default is
non-deterministic, only used when no BAM index is provided
--sample-region <SAMPLE_REGION>
Specify a region for sampling reads from when estimating the threshold
probability. If this option is not provided, but --region is provided,
the genomic interval passed to --region will be used. Format should be
<chrom_name>:<start>-<end> or <chrom_name>
--sampling-interval-size <SAMPLING_INTERVAL_SIZE>
Interval chunk size to process concurrently when estimating the
threshold probability, can be larger than the pileup processing
interval
[default: 1000000]
-p, --filter-percentile <FILTER_PERCENTILE>
Filter out modified base calls where the probability of the predicted
variant is below this confidence percentile. For example, 0.1 will
filter out the 10% lowest confidence modification calls
[default: 0.1]
--filter-threshold <FILTER_THRESHOLD>
Specify the filter threshold globally or per primary base. A global
filter threshold can be specified with by a decimal number (e.g.
0.75). Per-base thresholds can be specified by colon-separated values,
for example C:0.75 specifies a threshold value of 0.75 for cytosine
modification calls. Additional per-base thresholds can be specified by
repeating the option: for example --filter-threshold C:0.75
--filter-threshold A:0.70 or specify a single base option and a
default for all other bases with: --filter-threshold A:0.70
--filter-threshold 0.9 will specify a threshold value of 0.70 for
adenine and 0.9 for all other base modification calls
--mod-threshold <MOD_THRESHOLDS>
Specify a passing threshold to use for a base modification,
independent of the threshold for the primary sequence base or the
default. For example, to set the pass threshold for 5hmC to 0.8 use
`--mod-threshold h:0.8`. The pass threshold will still be estimated as
usual and used for canonical cytosine and other modifications unless
the `--filter-threshold` option is also passed. See the online
documentation for more details
--no-filtering
Don't filter base modification calls, assign each base modification to
the highest probability prediction
--edge-filter <EDGE_FILTER>
Discard base modification calls that are this many bases from the
start or the end of the read. Two comma-separated values may be
provided to asymmetrically filter out base modification calls from the
start and end of the reads. For example, 4,8 will filter out base
modification calls in the first 4 and last 8 bases of the read
--invert-edge-filter
Invert the edge filter, instead of filtering out base modification
calls at the ends of reads, only _keep_ base modification calls at the
ends of reads. E.g. if usually, "4,8" would remove (i.e. filter out)
base modification calls in the first 4 and last 8 bases of the read,
using this flag will keep only base modification calls in the first 4
and last 8 bases
--motif <MOTIF> <MOTIF>
Filter out any base modification call that isn't part of a basecall
sequence motif This argument can be passed multiple times. Format is
<motif_sequence> <offset>. For example the argument to match CpG
dinucleotides is `--motif CG 0`, or to match CG[5mC]G the argument
would be `--motif CGCG 2`
--cpg
Shorthand for --motif CG 0
--discard-motifs
Discard base modification calls that match the provided motifs
(instead of keeping them)
--output-sam
Output SAM format instead of BAM
-h, --help
Print help (see a summary with '-h')
modbam repair
Repair MM and ML tags in one bam with the correct tags from another. To use this
command, both modBAMs _must_ be sorted by read name. The "donor" modBAM's reads
must be a superset of the acceptor's reads. Extra reads in the donor are
allowed, and multiple reads with the same name (secondary, etc.) are allowed in
the acceptor. Reads with an empty SEQ field cannot be repaired and will be
rejected. Reads where there is an ambiguous alignment of the acceptor to the
donor will be rejected (and logged). See the full documentation for details
Usage: modkit modbam repair [OPTIONS] --donor-bam <DONOR_BAM> --acceptor-bam <ACCEPTOR_BAM> --output-bam <OUTPUT_BAM>
Options:
-d, --donor-bam <DONOR_BAM>
Donor modBAM with original MM/ML tags. Must be sorted by read name
-a, --acceptor-bam <ACCEPTOR_BAM>
Acceptor modBAM with reads to have MM/ML base modification data
projected on to. Must be sorted by read name
-o, --output-bam <OUTPUT_BAM>
output modBAM location
--log-filepath <LOG_FILEPATH>
File to write logs to, it is recommended to use this option as some
reads may be rejected and logged here
-t, --threads <THREADS>
The number of threads to use [default: 4]
-h, --help
Print help
modbam check-tags
Usage: modkit modbam check-tags [OPTIONS] <IN_BAM>
Arguments:
<IN_BAM>
Input modBam, can be a path to a file or one of `-` or `stdin` to
specify a stream from standard input
Options:
--permissive
Don't exit 1 when invalid records are found in the input
-h, --help
Print help (see a summary with '-h')
IO Options:
-o, --out-dir <OUT_DIR>
Write output tables into this directory. The directory will be created
if it doesn't exist
-f, --force
Force overwrite of previous output
--prefix <PREFIX>
Prefix output files with this string
Compute Options:
-t, --threads <THREADS>
Number of threads to use
[default: 4]
--ignore-index
Perform a linear scan of the modBAM even if the index is found
-i, --interval-size <INTERVAL_SIZE>
When using regions, interval chunk size in base pairs to process
concurrently. Smaller interval chunk sizes will use less memory but
incur more overhead
[default: 5000000]
Logging Options:
--log-filepath <LOG_FILEPATH>
Specify a file for debug logs to be written to, otherwise ignore them.
Setting a file is recommended
--suppress-progress
Hide the progress bar
Selection Options:
-n, --num-reads <NUM_READS>
Approximate maximum number of reads to use, especially recommended
when using a large BAM without an index. If an indexed BAM is
provided, the reads will be sampled evenly over the length of the
aligned reference. If a region is passed with the --region option,
they will be sampled over the region. Actual number of reads used may
deviate slightly from this number
--allow-non-primary
Check tags on non-primary alignments as well. Keep in mind this may
incur a double-counting of the read with its primary mapping
--mapped-only
Only check alignments that are mapped
--region <REGION>
Process only the specified region of the BAM when collecting
probabilities. Format should be <chrom_name>:<start>-<end> or
<chrom_name>
--head <HEAD>
Use the first N reads without sampling. Shorthand for --ignore-index
--num-reads N
Modkit Installation for macOS
Prerequisites: Apple Silicon Mac, macOS 11.0+
MPS GPU builds only: macOS 12.3+ (Monterey or later) is required when
MODKIT_MPS_SUPPORT=1. The script exits early on older systems for MPS builds only. Builds without MPS (MODKIT_MPS_SUPPORT=0, the default) run on macOS 11.0+.
Quick Start
# Without MPS GPU support (default)
bash mac_compile_modkit.sh ~/tools
# With MPS GPU support (requires Python + PyTorch)
MODKIT_MPS_SUPPORT=1 bash mac_compile_modkit.sh ~/tools
The default (no-MPS) build installs modkit without Python or PyTorch. Takes 5–10 minutes. The MPS build adds Metal GPU acceleration and takes 10–15 minutes.
What Gets Installed
Always installed:
- Xcode Command Line Tools, Homebrew, Rust & Cargo
- Compiled modkit binary at
~/tools/modkit/target/release/modkit
Additionally installed when MODKIT_MPS_SUPPORT=1:
- Python virtual environment with PyTorch (GPU-enabled via Metal Performance Shaders)
After Installation
The installer automatically adds the environment setup to ~/.zprofile, so modkit is available in every new terminal session silently — no output is printed on shell startup.
To activate in the current session immediately after install:
source ~/tools/setup_modkit_env.sh ~/tools
modkit --version
To see full environment details:
source ~/tools/setup_modkit_env.sh ~/tools --verbose
setup_modkit_env.sh configures:
PATH— so you can typemodkitdirectlyLIBTORCH,DYLD_LIBRARY_PATH,LD_LIBRARY_PATH— only set for MPS builds; required for the binary to find libtorch
MPS GPU Support
MODKIT_MPS_SUPPORT=1 enables Metal Performance Shaders GPU acceleration. This requires Python and PyTorch, which the script installs automatically.
| Variable | Values | Default | Purpose |
|---|---|---|---|
MODKIT_MPS_SUPPORT | 0, 1 | 0 | Enable MPS GPU support |
Python Version Control
These variables are only used when
MODKIT_MPS_SUPPORT=1.
Three environment variables control Python selection:
| Variable | Values | Default | Purpose |
|---|---|---|---|
MODKIT_PYTHON_PROVIDER | auto, system, pyenv, uv | auto | Which Python manager to use |
MODKIT_PYTHON_VERSION | e.g. 3.11.9, 3.12 | (empty) | Request a specific Python version |
MODKIT_USE_UV | auto, 0, 1 | auto | Use uv for venv and pip operations |
Provider Notes
auto: UsesuvorpyenvifMODKIT_PYTHON_VERSIONis set and they are available; otherwise falls back to systempython3. If a version is requested but neither tool is available, the script warns and prompts before falling back.system: Usespython3from$PATH. Version cannot be controlled.pyenv: Installs the requested version if needed. Does not change your global pyenv version.uv: Fastest option. Installs Python and manages the venv/pip. Auto-installed via Homebrew if needed.
Venv reuse: If a virtual environment already exists from a different Python configuration, the script detects the mismatch and prompts you to recreate it.
Examples
# No MPS (default): no Python or PyTorch needed
bash mac_compile_modkit.sh ~/tools
# MPS build with system Python
MODKIT_MPS_SUPPORT=1 bash mac_compile_modkit.sh ~/tools
# MPS build, specific Python version via pyenv
MODKIT_MPS_SUPPORT=1 MODKIT_PYTHON_PROVIDER=pyenv MODKIT_PYTHON_VERSION=3.11.9 bash mac_compile_modkit.sh ~/tools
# MPS build, specific Python version via uv (fastest)
MODKIT_MPS_SUPPORT=1 MODKIT_PYTHON_PROVIDER=uv MODKIT_PYTHON_VERSION=3.12 bash mac_compile_modkit.sh ~/tools
# MPS build, force standard pip (no uv)
MODKIT_MPS_SUPPORT=1 MODKIT_USE_UV=0 bash mac_compile_modkit.sh ~/tools
# Specific modkit version (no MPS)
bash mac_compile_modkit.sh ~/tools v0.5.0
# Specific modkit version (with MPS)
MODKIT_MPS_SUPPORT=1 bash mac_compile_modkit.sh ~/tools v0.5.0
Common Issues & Fixes
“Could not resolve a usable Python executable”
Only relevant for
MODKIT_MPS_SUPPORT=1builds.
python3 --version # check if Python is available
brew install python@3.11 # install if missing
“PyTorch verification failed”
Only relevant for
MODKIT_MPS_SUPPORT=1builds.
source ~/tools/setup_modkit_env.sh ~/tools
~/tools/venv_modkit/bin/python -m pip install --upgrade pip torch numpy
“Compilation failed”
No-MPS build:
cd ~/tools/modkit
cargo clean
cargo build --release --features accelerate
MPS build:
Always source the environment before rebuilding. Without it,
LIBTORCHis unset and the build will fail again.
source ~/tools/setup_modkit_env.sh ~/tools
cd ~/tools/modkit
cargo clean
cargo build --release --features accelerate,tch
“MPS available: false”
MPS support is opt-in (MODKIT_MPS_SUPPORT=1). If you built without MPS, this is expected — rebuild with MODKIT_MPS_SUPPORT=1 to enable GPU acceleration.
For an MPS build where GPU is still unavailable:
- Confirm Apple Silicon:
uname -mshould printarm64 - Confirm macOS 12.3+:
sw_vers -productVersion - CPU-only fallback is used automatically if MPS is unavailable
Help & Details
bash mac_compile_modkit.sh --help # all options
cat ~/tools/installation_info.txt # paths, versions, reproducible commands
modkit --help # modkit usage
Troubleshooting
It’s recommended to run all modkit commands with the --log-filepath <path-to-file>
option set. When unexpected outputs are produced inspecting this file will often indicate
the reason.
Missing secondary and supplementary alignments in output
As of v0.2.4 secondary and supplementary alignments are supported in adjust-mods, update-tags, call-mods, and (optionally) in extract.
However, in order to use these alignment records correctly, the MN tag must be present and correct in the record.
The MN tag indicates the length of the sequence corresponding to the MM and ML tags.
As of dorado v0.5.0 the MN tag is output when modified base calls are produced.
If the aligner has hard-clipped the sequence, this number will not match the sequence length and the record cannot be used.
Similarly, if the SEQ field is empty (sequence length zero), the record cannot be used.
One way to use supplementary alignments is to specify the -Y flag when using dorado or minimap2.
For these programs, when -Y is specified, the sequence will not be hardclipped in supplementary alignments and will be present in secondary alignments.
Other mapping algorithms that are “MM tag-aware” may allow hard-clipping and update the MM and ML tags, modkit will accept these records as long as the MN tag indicates the correct sequence length.
No rows in modkit pileup output.
First, check the logfile, there may be many lines with a variant of
record 905b4cb4-15e2-4060-9c98-866d0aff78bb has un-allowed mode
(ImplicitProbModified), use '--force-allow-implicit' or 'modkit update-tags --mode
ambiguous
As suggested, using update or setting the
--force-allow-implicit flag should produce output from these records.
If the MM tag contains an un-supported mod-code (a limitation that will be removed in a future release), these errors will be logged. To process the modBAM, first convert the tags.
In general, if there are MM/ML tags in the modBAM and the bedMethyl is still empty the log file will contain a line explaining why each record is being skipped.
Not sampling enough reads to estimate threshold.
If you have previously downsampled the modBAM to a specific region, for example with a command like
samtools view -bh input.sorted.bam chr1
modkit will still try and sample reads evenly across the entire genome but fail to find
any aligned to other contigs. To remedy this, either pass the same --region chr1 option
or set the --sampling-frac 1.0. The former will set modkit to only use the region
present in the BAM. For example,
modkit pileup input.bam output.bed --region chr1
# or
modkit pileup input.bam output.bed --sample-region chr1
The latter will sample all of the reads in the BAM, which may slow
down the process, so in general the best advice is to either use the same --region <contig> when using modkit or don’t downsample the BAM ahead of time (and use --region <contig> in order to get the desired bedMethyl).
The summary, sample-probs, and pileup subcommands all use the same --region
option.
CG positions are missing from output bedMethyl.
When running with --preset traditional or --cpg the resultant bedMethyl file will only
contains CG positions. However, it will not include positions for which the pass coverage
is zero (see the column
descriptions). This is to be
expected.
Frequently asked questions
How are base modification probabilities calculated?
Base modifications are assigned a probability reflecting the confidence the base modification detection algorithm has in making a decision about the modification state of the molecule at a particular position.
The probabilities are parsed from the ML tag in the BAM record. These values reflect the probability of the base having a specific modification, modkit uses these values and calculates the probability for each modification as well as the probability the base is canonical:
\[
P_{\text{canonical}} = 1 - \sum_{m \in \textbf{M}} P_{m}
\]
where \(\textbf{M}\) is the set of all of the potential modifications for the base.
For example, consider using a m6A model that predicts m6A or canonical bases at adenine residues, if the \( P_{\text{m6A}} = 0.9 \) then the probability of canonical \( \text{A} \) is \( P_{\text{canonical}} = 1 - P_{\text{m6A}} = 0.1 \). Or considering a typical case for cytosine modifications where the model predicts 5hmC, 5mC, and canonical cytosine:
\[ P_{\text{5mC}} = 0.7, \\ P_{\text{5hmC}} = 0.2, \\ P_{\text{canonical}} = 1 - P_{\text{5mC}} + P_{\text{5hmC}} = 0.1, \\ \]
A potential confusion is that modkit does not assume a base is canonical if the probability of modification is close to \( \frac{1}{N_{\text{classes}}} \), the lowest probability the algorithm may assign.
What value for --filter-threshold should I use?
The same way that you may remove low quality data as a first step to any processing, modkit will filter out the lowest confidence base modification probabilities.
The filter threshold (or pass threshold) defines the minimum probability required for a read’s base modification information at a particular position to be used in a downstream step.
This does not remove the whole read from consideration, just the base modification information attributed to a particular position in the read will be removed.
The most common place to encounter filtering is in pileup, where base modification probabilities falling below the pass threshold will be tabulated in the \( \text{N}_{\text{Fail}} \) column instead of the \( \text{N}_{\text{valid}} \) column.
For highest accuracy, the general recommendation is to let modkit estimate this value for you based on the input data.
The value is calculated by first taking a sample of the base modification probabilities from the input dataset and determining the \(10^{\text{th}}\) percentile probability value.
This percentile can be changed with the --filter-percentile option.
Passing a value to --filter-threshold and/or --mod-threshold that is higher or lower than the estimated value will have the effect of excluding or including more probabilities, respectively.
It may be a good idea to inspect the distribution of probability values in your data, the modkit sample-probs command is designed for this task.
Use the --hist and --out-dir options to collect a histogram of the prediction probabilities for each canonical base and modification.
Current limitations
Known limitations and forecasts for when they will be removed.
- During
modkit pileup, it is assumed that each read should only have one primary alignment. If a read has more than one primary alignment it will be double counted, it is therefore recommended to remove duplicates before running Modkit. - The MAP-based p-value metric (details) performs a test that there is a difference in modification (of any kind) between two conditions. If a position has multiple base modification calls (such as 5hmC and 5mC) the calls are summed together into a single “modified” count. If a position differs only in the modification type (such as one condition has more 5hmC and the other has more 5mC) this effect will not be captured in the MAP-based p-value. The likelihood ratio test does capture changes in modification type.
- The MAP-based p-value is not available when performing DMR on regions. This is because there is potentially large variability in the number of modified bases and coverage over regions. This variability translates into varying degrees of statistical power and makes comparisons difficult to interpret.
- In Modkit versions v0.6.0 and v0.6.1
pileupwill saturate depth at 65,535 (maximum for a unsigned 16-bit integer). If your modBAM has a depth greater than this value, it is recommended to use the--high-depthflag so that 65,535 reads will be used at each genomic position. To achieve greater depth, divide the modBAM into subsets with depth <= 65,535, runpileupon each, thenmodbam merge. This limitation will be removed in the future when it can be proven not to have any performance or resource regression.
Performance considerations
Sharding a large modBAM by region.
The --region option in pileup, summary, and sample-probs can be used to
operate on a subset of records in a large BAM. If you’re working in a
distributed environment, the genome could be sharded into large sections which
are specified to modkit in concurrent processes and merged afterward in a
“map-reduce” pattern.
Setting the --interval-size and --chunk-size.
Whenever operating on a sorted, indexed BAM, modkit will operate in parallel on disjoint spans of the genome.
The length of these spans (i.e. intervals) can be determined by the --interval-size or the --sampling-interval-size (for the sampling algorithm only).
The defaults for these parameters works well for genomes such as the human genome usually at CpG contexts.
For smaller genomes with high coverage, you may decide to decrease the interval size in order to take advantage of parallelism.
When using transcriptomes multiple reference transcripts will be grouped - extra parameters are not required.
The pileup subcommand also has a --chunk-size option that will limit the total number of intervals computed on in parallel.
By default, modkit will set this parameter to be 50% larger than the number of threads.
In general, this is a good setting for balancing parallelism and memory usage.
Increasing the --chunk-size can increase parallelism (and decrease run time) but will consume more memory.
One note is that using “all-context” base modification models where a base modification call may be at every cytosine, adenine, or both, will require more memory than CpG models.
In these cases, modkit pileup must simply hold more base modification records in memory for a given interval-size.
It is possible to reduce memory usage (usually at no run-time cost) by decreasing the --interval-size parameter.
Memory usage in modkit extract.
Transforming reads into a table with modkit extract can produce large files (especially with long reads).
Passing the --bgzf flag will produce compressed (via gzp) outputs and can dramatically reduce disk usage.
Before the data can be written to disk, however, it is enqueued in memory and can potentially create a large memory burden.
There are a few ways to decrease the amount of memory modkit extract will use in these cases:
- Lower the
--queue-size, this decreased the number of batches that will be held in flight. - Use
--ignore-indexthis will forcemodkit extractto run a serial scan of the mod-BAM. - Decrease the
--interval-size, this will decrease the size of the batches. - Use
--include-bedon regions of interest to cull reads overlapping only regions. - Use
--motifand/or--ignore-implicitto reduce the number of records written per read.
Parallelism in motif search, motif refine, and when to --skip-search
The search algorithm takes advantage of parallelism at nearly every step and therefore hugely benefits from running with as many threads as possible (specified with --threads).
This horizontal scalability is most easily seen in the secondary search step where (by default) 129536 individual “seed sequences” are evaluated for potential refinement.
If you find that this search is taking a very long time (indicated by the progress bar message “<mod_code> seeds searched”) you may consider one of the following:
- Increase the
--exhaustive-seed-min-log-oddsparameter, this will decrease the number of seeds passed on to the refinement step (which is more computationally expensive). - Decrease the
--exhaustive-seed-lento 2 or decrease the--context-size, this will exponentially decrease the number of seeds to be searched.
You may also decide to run --skip-search first and inspect the results.
Algorithm details
- Filtering low confidence base modification calls
- Removing and combining modified base types
- Ignoreing and combining base modification calls
- Differential methylation scoring and differential modification p-values
Partitioning pass and fail base modification calls.
Base modification calls can be removed if they are low confidence based on the predicted modification probabilities.
In general, modkit will estimate a pass confidence threshold value based on the input data.
Threshold values for modifications on a primary sequence base can be specified on the command line with the --filter-threshold option.
For example to set a threshold for cytosine modifications at 0.8 and adenine modifications at 0.9 provide --filter-threshold C:0.8 --filter-threshold A:0.9.
Pass threshold values per base modification can also be specified.
For example, to specify a threshold for canonical adenine at 0.8 and 6mA at 0.9 use --filter-threshold A:0.8 --mod-thresholds a:0.9.
Or to specify a threshold of 0.8 for 5mC, 0.9 for 5hmC, and 0.85 for canonical cytosine: --filter-threshold C:0.85 --mod-threshold m:0.8 --mod-threshold h:0.9
Keep in mind that the --mod-threshold option will treat A, C, G, and T and “any-mod” as per the specification.
Further details
- Examples of how thresholds affect base modification calls.
- Numerical details of how thresholds are calculated on the fly.
Examples of how thresholds affect base modification calls
The following examples are meant to demonstrate how individual base modification calls will be made during
pileup or call-mods. Important to remember that the probability of the modification needs to be >= the
pass threshold value.
Two-way base modification calls
probability of canonical cytosine: p_C
probability of 5mC: p_m
threshold for all cytosine base modifications: threshold_C
{p_C: 0.25, p_m: 0.75}, threshold_C: 0.7 => call: 5mC (m), most likely.
{p_C: 0.25, p_m: 0.75}, threshold_C: 0.8 => call: Fail, both probabilities are below threshold.
Three-way base modification calls
probability of canonical cytosine: p_C
probability of 5mC: p_m
probability of 5hmC: p_h
threshold for all cytosine base modifications: threshold_C
{p_C: 0.05, p_m: 0.7, p_h: 0.25}, threshold_C: 0.7 => call: 5mC (m), most likely.
{p_C: 0.15, p_m: 0.6, p_h: 0.25}, threshold_C: 0.7 => Fail, all below threshold.
Three-way base modification calls with modification-specific thresholds
probability of canonical cytosine: p_C
probability of 5mC: p_m
probability of 5hmC: p_h
threshold for all canonical cytosine: mod_threshold_C
threshold for all 5mC: mod_threshold_m
threshold for all 5hmC: mod_threshold_h
command line: --filter-threshold C:0.7 --mod-threshold m:0.8 --mod-threshold h:0.9
filter_threshold C: 0.7
mod_threshold_m: 0.8
mod_threshold_h: 0.9
{p_C: 0.05, p_m: 0.85, p_h: 0.1} => call: 5mC (m) most likely
filter_threshold C: 0.7
mod_threshold_m: 0.8
mod_threshold_h: 0.9
{p_C: 0.75, p_m: 0.05, p_h: 0.2} => call: C (canonical) most likely
{p_C: 0.05, p_m: 0.75, p_h: 0.2} => Fail, all below respective thresholds
{p_C: 0.1, p_m: 0.05, p_h: 0.85} => Fail, all below respective thresholds
Filtering base modification calls.
Modified base calls are qualitied with a probability that is contained in the ML tag (see the
specification). We calculate the confidence that the model
has in the base modification prediction as \(\mathcal{q} = argmax(\textbf{P})\) where \(\textbf{P}\) is the
vector of probabilities for each modification. For example, given a model that can classify canonical
cytosine, 5mC, and 5hmC, \(\textbf{P}\) is \([P_{C}, P_m, P_h]\), and \(\mathcal{q}\) will be \(\mathcal{q} =
argmax(P_{C}, P_m, P_h)\), the maximum of the three probabilities. Filtering in modkit is performed by
first determining the value of \(\mathcal{q}\) for the lowest n-th percentile of calls (10th percentile by
default). The threshold value is typically an estimate because the base modification probabilities are
sampled from a subset of the reads. All calls can be used to calculate the exact value, but in practice the
approximation gives the same value. Base modification calls with a confidence value less than this number will
not be counted. Determination of the threshold value can be performed on the fly (by sampling) or the
threshold value can be specified on the command line with the --filter_threshold flag. The sample-probs
command can be used to quickly estimate the value of \(\mathcal{q}\) at various percentiles.
A note on the “probability of modification” (.) MM flag.
The . flag (e.g. C+m.) indicates that primary sequence bases without base modification calls can be inferred to be
canonical (SAM tags). Some base modification callers,
for example dorado have a default threshold, below which a base modification
probability will be omitted (meaning it will be inferred to be a canonical/unmodified base). In general, omitting
the base modification probabilities when they are very confidently canonical will not change the results from modkit.
However, since the base modification probabilities are effectively changed to 100% canonical, can affect the
estimation of the pass threshold.
DMR model and scoring details (dmr pair and dmr multi)
Likelihood ratio scoring details
The aim of modkit dmr is to enable exploratory data analysis of methylation patterns. To that aim, the approach to
scoring methylation differences is intended to be simple and interpretable. For every region provided, within a sample,
we model each potentially methylated base as arising from the same distribution. In other words, we discard the relative
ordering of the base modification calls within a region. We then define a model for the frequency of observing each base
modification state. In the case of methylated versus unmodified (5mC vs C, or 6mA vs A), we use the binomial distribution
and model the probability of methylation \(p\) as a beta-distributed random variable:
\[ \mathbf{X}|p \sim \text{Bin}(n, p) \] \[ p \sim \text{Beta}(\alpha, \beta) \]
where \(n\) is the number of potentially methylated bases reported on in the region and \(\mathbf{X}\) is the vector of counts (canonical and methylated).
In the case where there are more than two states (for example, 5hmC, 5mC, and unmodified C) we use a multinomial distribution and a Dirichlet as the base distribution: \[ \mathbf{X}|\pi \sim \text{Mult}(n, \pi) \]
\[ \pi \sim \text{Dir}(\alpha) \]
Let \(\theta\) be the parameters describing the posterior distribution ( \( \alpha, \beta \) for the binary case,
and \(\alpha \) in the general case). The score reported is the result of the following log marginal likelihood
ratio :
\[ \text{score} = \text{log}(\frac{l_{\theta_{a}}( \mathbf{X_a} ) l_{\theta_{b}} ( \mathbf{X_b} )}{l_{\theta_{a+b}} (\mathbf{X_{a+b}} )}) \]
Where \(\theta_a\) and \(\theta_b\) are the posterior distributions with the two conditions modeled separately, and \(\theta_{a+b}\) is the posterior when the two conditions are modeled together. The function \(l_{\theta}(\mathbf{X}) \) is the log marginal likelihood of the counts under the parameters of the model \(\theta\). For all cases, we use Jeffrey’s prior as the prior distribution.
MAP-based p-value
This metric models the effect size (i.e. the difference) in base modification (of any kind) between two conditions. For example, if one condition has 8 of 10 reads reporting modification, 80%, and the other condition has 2 of 10, 20%, then the effect size 0.6 or 60%. This metric only captures changes in modified versus unmodified bases, in other words, changes in modification type will not be captured by this metric. See the limitations for details. The DMR module in modkit uses Bernoulli trials (modified/not-modified) and a prior to calculate a posterior distribution over \(p\), the true probability that the site is modified. The posterior distribution over \(p\) given some observations, \(X\), is \(P(p | X)\), is a Beta distribution. Where \(X\) is the observations (\(N_{\text{mod}}\) and \(N_{\text{canonical}}\)), the number of reads calling a modified base and a canonical base, respectively.
\[ P(p | X) = \text{Beta}(\alpha_0 + N_{\text{mod}}, \beta_0 + N_{\text{can}}) \] Where \(\alpha_0\) and \(\beta_0\) are the parameters for the prior distribution \(\text{Beta}(\alpha_0, \beta_0)\). The advantage to this model is that as you collect more coverage, the variance of the posterior gets smaller - you’re more confident that the true value of \(p\) is near the empirical mean. But when you have low coverage, you keep the uncertainty around.
More formally, let \(\textbf{X}\) be a Beta-distributed random variable representing the posterior distribution over \(p\) for the first condition and \(\textbf{Y}\) is the same for the second condition. Finally, let \(f(x)\) be the probability density of the difference \(x\) in \(\textbf{X}\) and \(\textbf{Y}\). Then the MAP-based p-value, \(p_{\text{MAP}}\), is the posterior odds of the effect size being zero (no difference) versus the maxumum a posteriori (MAP) outcome:
\[ p_{\text{MAP}} = \frac{f(0.0)}{f(x_{\text{MAP}})} \ \] \[ f(x) = PDF_{\textbf{Z}}(x) \ \] \[ \textbf{Z} = \textbf{X} - \textbf{Y} \ \] \[ \textbf{X} \sim \text{Beta}(\alpha_1, \beta_1) \ \] \[ \textbf{Y} \sim \text{Beta}(\alpha_2, \beta_2) \ \]
This metric was proposed by Makowski et al. and can be easily visualized. Consider an example with a true effect size of 0.8 at two coverages, 5 reads and 10 reads. For an effect size of 0.8, the \(p\) for the low modification condition and the high modification condition is 0.1 and 0.9, respectively. This corresponds to 4 of 5 reads being called methylated in the low-coverage case, and 9 of 10 reads being called modified in the high-coverage condition. The reciprocal counts are used in both conditions, so 1 of 5 for the low-coverage and 1 of 10 for the high-coverage.
- Low coverage = 5: 4 of 5 modified versus 1 of 5 modified
- High coverage = 10: 9 of 10 modified versus 1 of 10 modified
Starting with a prior of \(\text{Beta}(0.5, 0.5)\), we can calculate the posterior density for the two conditions:
What we need to calculate is the probability distribution of the difference (the effect size) between the two conditions (high-modification and low-modification). This distribution can be done using a piecewise solution described by Pham-Gia, Turkkan, and Eng in 1993, the distribution is shown below:
The MAP-based p-value is the ratio of the circles over the triangles.
The implementation in modkit takes a small short-cut however, and uses the empirical effect size \(d = \hat{p}_1 - \hat{p}_2\) instead of the maximum a posteriori outcome.
\[ \text{p-MAP}^{\prime} = \frac{f(0.0)}{f(d)} \ \]
\[ d = \hat{p}_1 - \hat{p}_2 \ \]
\[ \hat{p} = \frac{ N_{\text{mod}} }{ N_{\text{canonical}} } \ \]
DMR segmentation hidden Markov model
When performing “single-site” analysis with modkit dmr pair (by omitting the --regions option) you can optionally run the “segmentation” model at the same time by passing the --segment option with a filepath to write the segments to.
The model is a simple 2-state hidden Markov model, shown below, where the two hidden states, “Different” and “Same” indicate that the position is either differentially methylated or not.

The model is run over the intersection of the modified positions in a pileup for which there is enough coverage, from one or more samples.
Transition parameters
There are two transition probability parameters, \(p\) and \(d\).
The \(p\) parameter is the probability of transitioning to the “Different” state, and can be roughly though of as the probability of a given site being differentially modified without any information about the site.
The \(d\) parameter is the maximum probability of remaining in the “Different” state, it is a maximum because the value of \(d\) will change dynamically depending on the proximity of the next modified site.
The model proposes that modified bases in close proximity will share modification characteristics.
Specifically, when a site is differentially modified the probability of the next site also being differentially modified depends on how close the next site happens to be.
For example, if a CpG dinucleotide is differentially modified and is immediately followed by another CpG (sequence is CGCG) we have the maximum expectation that the following site is also differentially modified.
However, as the next site becomes farther away (say the next site is one thousand base pairs away, CG[N x 1000]CG) these sites are not likely correlated and the probability of the next site being differentially modified decreases towards \(p\).
The chart below shows how the probability of staying in the “Different” state, \(d\), decreases as the distance to the next modified base increases.

In this case, the maximum value of \(d\) is 0.9, \(p\) is 0.1, and the decay_distance is 500 base pairs (these also happen to be the defaults).
This can be seen as the maximum value of both curves is 0.9, and the minimum value, reached at 500 base pairs, is 0.1.
These parameters can be set with the --diff-stay, --dmr-prior, and --decay-distance, parameters, respectively.
The two curves represent two different ways of interpolating the decay between the minimum (1) and the decay_distance, linear and logistic.
The --log-transition-decay flag will use the orange curve whereas the default is to use the blue curve.
In general, these settings don’t need to be adjusted.
However, if you want very fine-grained segmentation, use the --fine-grained option which will produce smaller regions but also decrease the rate at which sites are classified as “Different” when they are in fact not different.
Emission parameters
The emissions of the model are derived from the likelihood ratio score. One advantage to using this score is that differences in methylation type (i.e. changes from 5hmC to 5mC) will be modeled and detected. The score is transformed into a probability by \( p = e^{\text{-score}} \). The full description of the emission probabilities for the two states is:
\[ p_{\text{Same}} = e^{\text{-score}} \] \[ p_{\text{Different}} = 1 - p_{\text{same}} \]
Cohen’s h statistic for regions, single-sites, and segments
As the number of positions being compared becomes large, small effect sizes will become statistically significant with as measured by most tests. The MAP-based p-value calculation has a maximum coverage of 100 reads since it becomes unstable when the number of trials per condition grows beyond this number (essentially the p-value becomes zero). A similar problem is encountered when comparing regions of CpG or transcripts of m6As either provided as regions to compare or as discovered by the segmentation algorithm. To provide another metric that is more robust to high counts, Modkit DMR will output Cohen’s h statistic. Importantly this is a measure of changes in the proportion of modification of any type between the two conditions, similar to the MAP-based p-value, not a change in modification proportions (although it could be extended to measure this). In addition to the statistic, the high and low bound of the 95% confidence interval are reported. A CI value (high or low) of zero indicates that there is little certainty about there being a difference between the two conditions. Generally speaking, filtering or sorting on the lower bound is a good test for finding important changes.
Direct RNA Isoform DMR details
The RNA-specific DMR functions in Modkit use a Dirichlet-multinomial model. The model assumes that the underlying methylation-state proportions are drawn from a Dirichlet distribution: \[ p_i \sim \text{Dirichlet}(\alpha_{1}, \alpha_{2}, \dots, \alpha_{K}) \]
Where \( K \) is the number of methylation states (m6A and Inosine, for example).
The score is then calculated as the likelihood ratio statistic:
\[
\text{LRT} = 2\ (\text{log}\ L_{alt} - L_{null})
\]
Where the null model is one where all isoforms (or both conditions) share the same proportions and the alternative model is one where each isoform or condition has it’s own proportions.
The degrees of freedom is then calculated as
\[
df = (M - 1)(K - 1)
\]
where \( M \) is the number of isoforms (or \( 2 \) in the case of compare-tx-sites).
Finally, the p-value is calculated as
\[
p = P(\chi^2_{df}\ge\ LRT)
\]
Removing modification calls from BAMs.
There may be multiple possible base modifications to a specific canonical base.
For example, cytosine has at least four known modified variants. As
technologies are increasingly able to detect these chemical moieties, not all
downstream tools may be capable of accepting them, or you may want to convert
your data to make it comparable to other data with less specific resolution. To
address this issue, modkit implements a method for removing one or more DNA
base modification call from a BAM as well as a command line flag to simply
combine all modification calls when performing a pileup to generate a bedMethyl
file.
Removing DNA base modification probabilities.
BAM records containing probabilities for multiple base modifications at each residue can
be transformed to ignore one (or more) of the predicted base modifications. For example,
if a BAM file contains 5hmC and 5mC probabilities, the total probability is necessarily
distributed over 5mC, \(p_m\), 5hmC, \(p_h\), and canonical C, \(p_{C}\). modkit
implements a method, described below, for reducing the dimensionality of the probability
distribution by one or more of the predicted base modification classes.
Take for example a 3-way modification call for C, 5mC, and 5hmC, the probabilities of each being \(p_{C}\), \(p_{m}\), \(p_{h}\), respectively. To remove the 5hmC probability, \(p_{h}\) is distributed evenly across the other two options. In this example, the updates are \( p_C \leftarrow p_C + (\frac{p_h}{2}) \) and \( p_m \leftarrow p_m + (\frac{p_h}{2}) \).
Combining multiple base modifications into a single count.
Combine: --combine
In modkit the combine method can be used to produce binary modified/unmodified
counts. Continuing with the above example, the counts for number of modified reads,
N_mod, becomes the number of reads with 5hmC calls plus the number of reads with 5mC
calls. N_other_mod will always be 0. The raw_mod_code will be reported as the
ambiguous mod code for the canonical base. See
https://samtools.github.io/hts-specs/SAMtags.pdf for details on base modification codes
and schema.yaml in this project for details on the notation for counts.
Details on how reads are sampled with --num-reads and --sample-frac
Sampling approximately --num-reads from an aligned modBAM
This algorithm is meant be work quickly when the modBAM has generally uniform coverage over the aligned reference.
It works by dividing the reference into “intervals” of length --interval-size (or sometimes --sampling-interval-size is available as an override) and taking the first \( \textbf{N} \) reads from that interval.
The number of reads to take from that interval, \( \textbf{N} \), is determined by first proportionally distributing the number of requested reads over the contigs in the reference.
Then for each contig in the reference, the number of reads to be sampled from that contig are divided evenly over the intervals that tile that reference.
Here is a worked example:
contig_1, length = 100 contig_2, length = 50 contig_3, length = 10
Total reference length = 100 + 50 + 10 = 160
Requested reads: 1000 (--num-reads 1000)
contig_1 is \( \frac{100}{160}\) of the total, so it will get \( \lceil 1000 * \frac{100}{160} \rceil \).
A small “fudge factor” of 25% is added.
A table showing how many reads per interval and per contig is printed to the debug log (set with --log <file_name>).
The same arithmetic is done for contig_2 and contig_3.
After the number of reads per interval is calculated for each contig, Modkit will sample this many reads from the start of each interval:
contig_1 >------|-----|-----|-----|---<
^ ^ ^ ^ ^
N reads are taken from the start of each "^"
This algorithm is the “roughest” of the options, however, with a typical whole genome or whole transcriptome alignment provides a robust estimate.
Sampling --sampling-frac fraction of reads.
This is a more traditional option for sampling reads from the modBAM.
In this scenario each reader (more on that below) only keeps a fraction of the records for processing, and discards the others.
A “reader” can be a worker assigned to one interval of the reference (like above) or a single reader assigned to scan the entire modBAM file.
The latter is the case when no index is available (unaligned modBAM) or --ignore-index is passed.
The random state in the readers are seeded, so the same command will always sample the same records.
The following will change this determinism:
- No options or flags.
Deterministic unless the
--threadsor--interval-sizechanges. - Set
--seed, deterministic regardless of the number of workers (--threads), not deterministic when--interval-sizechanges!