Re-squiggle Algorithm¶
The electric current signal level data produced from a nanopore read is referred to as a squiggle. Base calling this squiggle information generally contains some errors compared to a reference sequence. The re-squiggle algorithm defines a new assignment from squiggle to reference sequence, hence a re-squiggle.
The re-squiggle algorithm is the basis for the Tombo framework. The re-squiggle algorithm takes as input a read file (in FAST5 format) containing raw signal and associated base calls. The base calls are mapped to a genome or transcriptome reference and then the raw signal is assigned to the reference sequence based on an expected current level model.
TL;DR:
The
tombo resquigglecomand must be run on a set of reads before modified base detection or other Tombo commands.A directory containing FAST5 read files and a genome/transcriptome reference must be provided.
The reference sequence may be previously known or discovered from this sample.
Importantly, the reference sequence is assumed to be correct, so polishing to create a personalized reference may improve performance, particularly for a divergent sample or poorly assembled reference.
Raw read FAST5 files must contain basecalls.
Add basecalls from a set of FASTQs to raw read files with the
tombo preprocess annotate_raw_with_fastqscommand.Read files need NOT contain
Eventsdata (as output withfast5mode from albacore).
Tombo currently only supports both DNA and RNA data (including R9.4 and R9.5; 1D and 1D2 data; R9.*.1 chemistries). Other data may produce sub-optimal results (e.g. R9.0 or R7 data).
DNA and RNA reads will be detected automatically and processed accordingly (set explicitly with
--dnaor--rna).Tombo does not perform spliced mapping. Thus a transcriptime reference must be passed to the re-squiggle command for RNA samples. For futher details on Tombo RNA processing see the RNA Processing section.
Run
tombo resquiggleover multiple cores with the--processesoption.
Algorithm Details¶
The re-squiggle algorithm occurs in five main steps described below.
Genome Mapping
Signal Normalization
Event Detection
Sequence to Signal Assignment
Resolve Skipped Bases
Genome Mapping¶
The genome mapping is performed via the python API to minimap2 (mappy python package).
Read base called sequence location within the FAST5 file is defined by the --basecall-group and --basecall-subgroups command line options. The default values of these parameters point to the default location for base calls from albacore or tombo preprocess annotate_raw_with_fastqs.
The genomic sequence for successfully mapped reads are then passed on to the Sequence to Signal Assignment stage.
Signal Normalization¶
Before the first iteration of the event detection and signal to sequence assignment steps, the raw signal for a read is normalized using median shift and MAD (median absolute deviation) scale parameters.
\(NormSignal = \frac{RawSignal - Shift}{Scale}\)
As of Tombo version 1.3 after the first iteration, new shift and scale parameters are computed by matching the expected signal levels with those observed from the first iteration of signal to seuquence assignment. The Theil-Sen estimator for the relationship between expected and observed signal levels is computed and used as a correction factor for the previous scale parameter. A shift correction factor is also computed taking the median of intercepts over each base in the read.
If either the shift or scale correction factors exceed a preset threshold, an additional round of sequence to signal assignment is performed. This continues until the corrections factors are small enough or a maximum number of iterations are performed. Command line parameters to control this procedure can be found using the tombo resquiggle --print-advanced-arguments command.
This method should be more robust to samples with higher modified base content than mean based sequence-dependent correction methods (e.g. M.O.M.).
This per-read sequence-dependent normalization has provided much better results than previous Tombo scaling methods and is thus strongly recommended. Previous scaling methods are still made available for research purposes (see tombo resquiggle --print-advanced-arguments).
Note
As of version 1.4, RNA samples are normalized over events after stall masking in order to provide more accurate normalzation factors. For RNA normalization compuation, the end of the read is trimmed (beginning in sequencing time), so that the DNA adapter does not effect normalization parameter estimation.
Event Detection¶
The Tombo resquiggle algorithm does not require the “Events” table (raw signal assignment to base calls). Instead, Tombo discovers events from the raw signal. This segmented signal makes downstream processing steps more efficient and stable. The Tombo event detection algorithm is different from the event detection performed in previous versions of albacore, but produces similar results.
Events are determined by identifying large shifts in current level, by taking the running difference between neighboring windows of raw signal (explicitly set this parameter with the --segmentation-parameters option). The largest jumps (or most significant via a t-test for RNA) are chosen as the breakpoints between events. The mean of normalized raw signal is then computed for each event.
The --segmentation-parameters values are optimized for DNA and RNA data types, so DNA and RNA read types should not be mixed in processing.
Note
For RNA samples, stalled bases are detected using a moving window mean approach and event boundaries located within a stalled base are removed from downstream processing.
Sequence to Signal Assignment¶
Given the mapped genomic sequence and normalized, segmented, raw signal, the sequence to signal assignment algorithm finds the most likely matching between these two.
This matching is found by a dynamic programming/dynamic time warpping algorithm to match event signal levels with expected signal levels given genomic sequence.
To compute this matching, first a static banded matrix is constructed by computing the z-score for event level (x-axis) against genomic positions (y-axis). The negative absolute value z-score is shifted to an expected value of zero to fill the banded matrix (see figure below). A forward pass computes the maximal cummulative score up to each matched event to genome position (see figure below).
At each iteration (moving from bottom left to top right) the maximal score is taken over three possibilities 1) staying in the same genomic position, and accumulating the shifted z-score 2) matching an event with a genomic position (with score bonus) 3) skipping this genomic position (with a score penalty). The score match and skip penalties are definied by the --signal-align-parameters option. The default values have been optimized for DNA and RNA data types. From this forward pass, the maximal score along the last genomic position is taken and traced back to obtain a matching of sequence and signal.
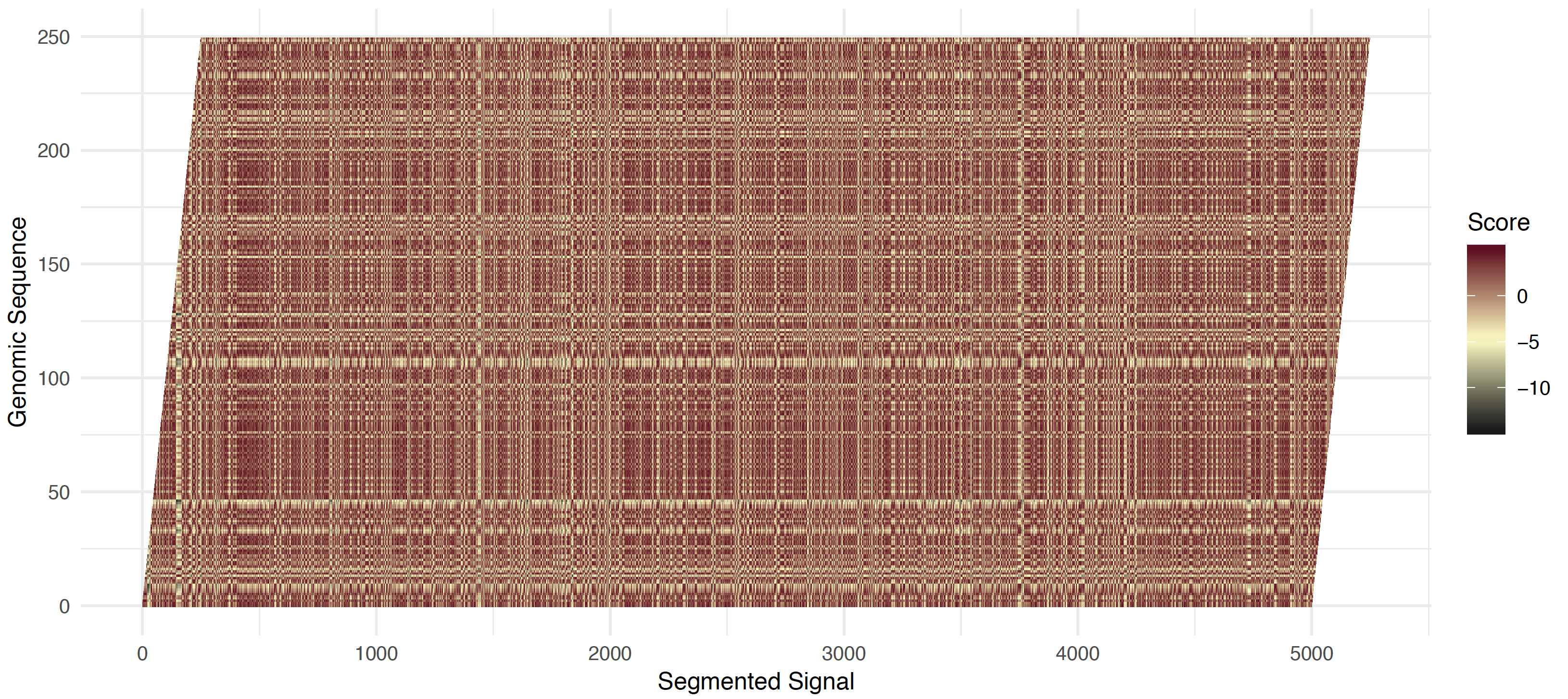
Read start shifted half-normal scores¶
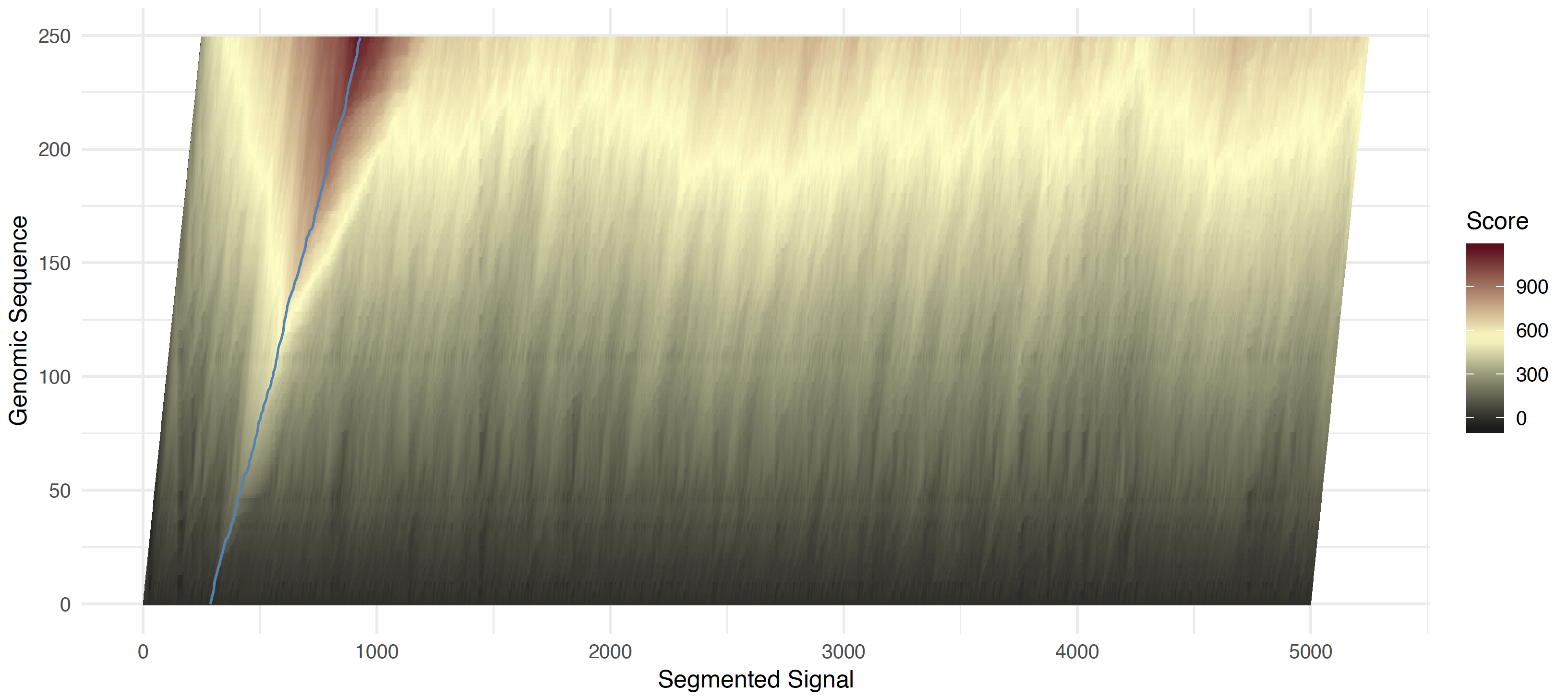
Read start forward pass scores and traceback path¶
The algorithm first uses a large bandwidth to identify the start of the genomic sequence within the events. This is necessary as some portion at the beginning of a read is not base called and some additional sequence may have been trimmed from the alignment.
If a read is short enough, then the whole sequence to signal matching will be performed with an appropriate static bandwidth.
For longer reads, the above computed geneome seqeunce start within the raw signal is taken and then the same dynamic programming solution is applied except a smaller adaptive band is now used (see figure below). The bandwidth is definied by the --signal-align-parameters option and again has been optimized for DNA and RNA data types. At each genomic position, the band position is defined to center on the maximal score of the forward pass from the previous base. This aims to ensure that the traceback path will remain within the adaptive window. There are edge cases where the valid matching leaves the adaptive band. These reads are filtered out and included in the failed read group Read event to sequence alignment extends beyond bandwidth.
Most reads can be processed with a smaller bandwidth, but if a read fails to be successfully re-squiggled a second, larger, “save” bandwidth is used to attempt to rescue a read and complete a successful sequence to signal assignment. For samples with many low quality reads, this can cause larger run times, but should speed up the vast majority of runs.
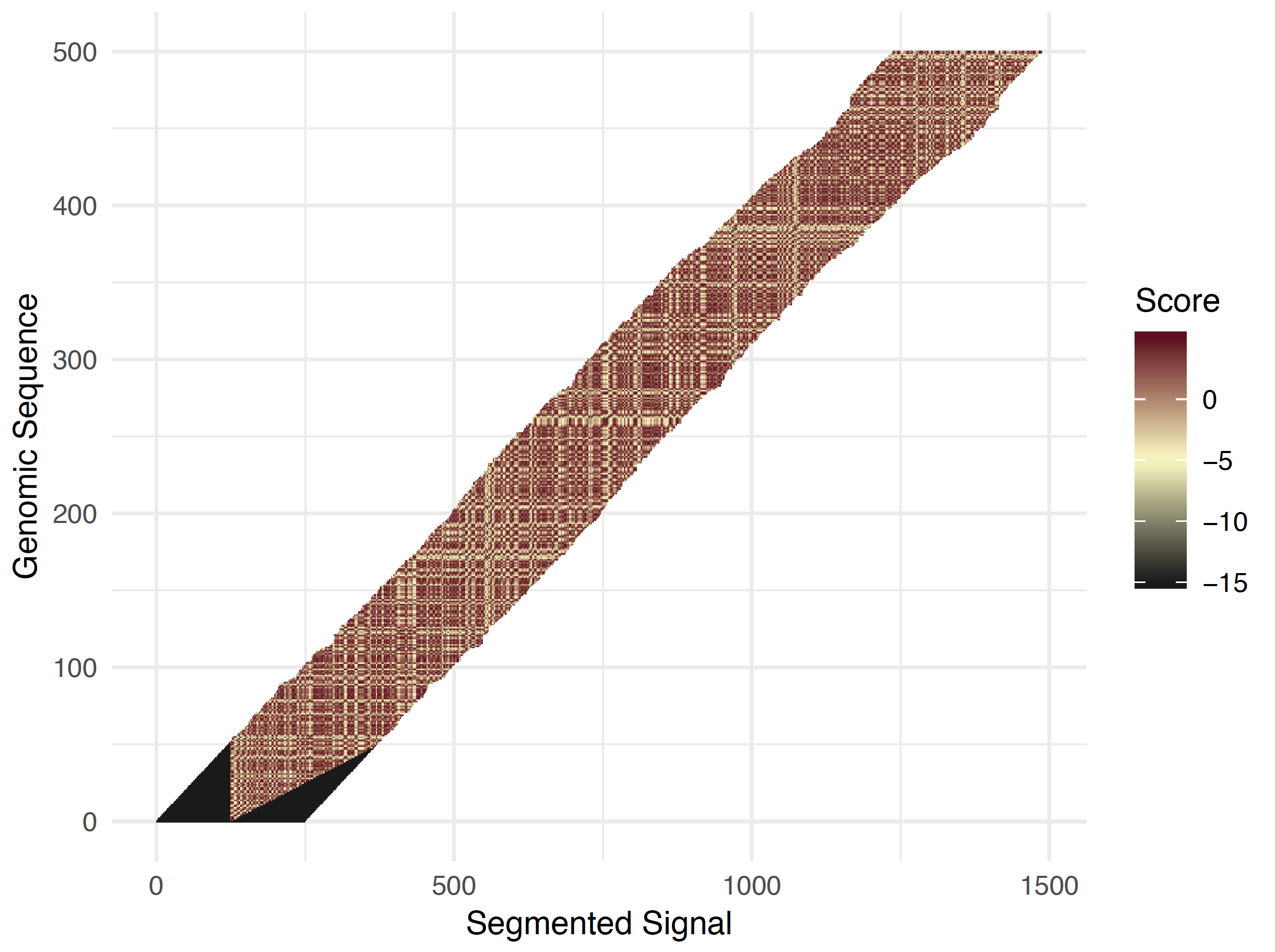
Full read adaptive banded shifted half-normal scores¶
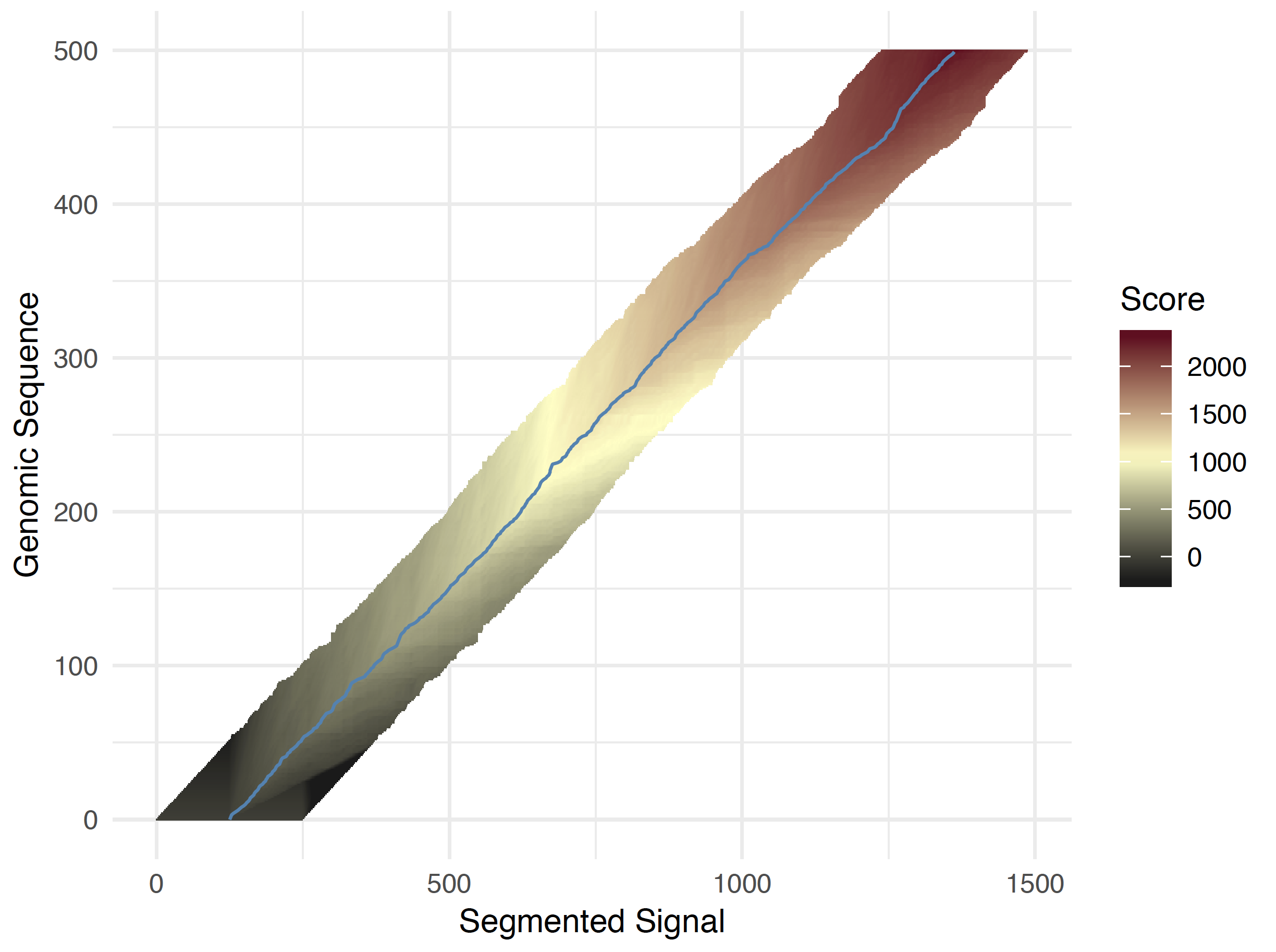
Full read adaptive banded forward pass scores¶
Resolve Skipped Bases¶
After the dynamic programming step, skipped bases must be resolved using the raw signal to obtain a matching of each genomic base to a bit of raw signal. A window around each skipped genomic base is identified. If a window does not contain enough raw signal to perform a raw signal search the window is expanded until enough signal is found. Overlapping windows are collapsed into a single window.
After deletion windows are identified, a dynamic programming algorithm very similar to the last step is performed. Importantly, the raw signal is used instead of events and the skip move is no longer allowed. Additionally, each genomic base is forced to contain a minimal number of raw observations to produce more robust assignments (explicitly set this value with the --segmentation-parameters option).
This completes the re-squiggle procedure producing a matching of a read’s raw signal to the mapped genomic sequence.
Common Failed Read Descriptions¶
Fastq slot not present in --basecall-group
Raw data is not found in Raw/Reads/Read_[read#]
These error indicates that a necessary peice of information for Tombo to run was not found in the FAST5 file. See
tombo preprocess annotate_raw_with_fastqsfor annotation of read files with basecalls in FASTQ format.
Alignment not produced
This error indicates that minimap2 (via mappy API) did not produce a valid mapping.
Could not close FAST5 file. Possibly corrupted file
This error indicates that an unexpected error occurred trying to open or close a read file. This can happen if the reads are being actively accessed by another program or if a file has been corrupted.
Read contains too many potential genomic deletions
Not enough raw signal around potential genomic deletion(s)
These errors indicate that the sequence to signal matching algorithm was unable to identify a valid path. This can occur if a sample contains sequence divergent from the provided reference sequence.
Poor raw to expected signal matching
This errors indicate that the dynamic programming algorithm produce a poorly scored matching of genomic sequence to raw signal (as definied by the
--signal-matching-score). Some potential sources for these errors include incorrect primary genomic mapping, incorrect genome sequence (compared to the biological sample), poor quality raw signal or an incompatible flowcell/library with included canonical models (only R9.5/4 flowcells currently supported; 2D reads are not supported; DNA and RNA are supported).
Unexpected error
This indicates that an error not expected by the Tombo re-squiggle algorithm has occured. This will result in a file containing the full error message for that read. Please report these bugs here.
Tombo FAST5 Format¶
The result of the re-squiggle algorithm writes the sequence to signal assignment back into the read FAST5 files (found in the --corrected-group slot; the default value is the default for all other Tombo commands to read in this data). When running the re-squiggle algorithm a second time on a set of reads, the --overwrite option is required in order to write over the previous Tombo results.
The --corrected-group slot contains attributes for the signal normalization (shift, scale, upper_limit, lower_limit, outlier_threshold and the tombo signal matching score) as well as a boolean flag indicating whether the read is DNA or RNA. Within the Alignment group, the gemomic mapped start, end, strand and chromosome as well as mapping statistics (number clipped start and end bases, matching, mismatching, inserted and deleted bases) are stored.
The Events slot contains a matrix with the matching of raw signal to genomic sequence. This slot contains a single attribute (read_start_rel_to_raw) giving the zero-based offset indicating the beginning of the read genomic sequence within the raw signal. Each entry in the Events table indicates the normalized mean signal level (norm_mean), optionally (triggered by the --include-event-stdev option) the normalized signal standard deviation (norm_stdev), the start position of this base (start), the length of this event in raw signal values (length) and the genomic base (base).
This information is accessed as needed for down-stream Tombo processing commands.
This data generally adds ~75% to the memory footprint of a minimal FAST5 file (containing raw and sequence data; NOT including a basecalling Events table). This may vary across files and sample types.
Important RNA note: Tombo only processes un-spliced mappings. As such, for potentially spliced transcripts a transcriptome reference file must be provided. While this makes Tombo RNA processing dependent upon a gene annotation, the transcriptome is a more appropriate setting for modified base detection. More details about RNA processing can be found in the RNA Processing section.
Minor RNA note: RNA reads pass through the pore in the 3’ to 5’ direction during sequencing. As such, the raw signal and albacore events are stored in the reverse direction from DNA reads. Tombo events for RNA data are stored in the opposite direction (corresponding to the genome sequence direction, not sequencing time direction) for several practical reasons. Thus if events are to be compared to the raw signal, the raw signal must be reversed. Tombo RNA models are stored in the same direction and thus may be considered inverted as compared to some other RNA HMM signal level models.
Tombo Index File¶
By default, Tombo will create a hidden file containing the essential genome mapping location for each validly processed read. This file will be located alongside the base fast5s directory. For example, if resquiggle is called on this directory /path/to/fast5/reads/ then the following file will be created /path/to/fast5/.reads.RawGenomeCorrected_000.tombo.index. This file should generally be about 1,000 times smaller than the corresponding FAST5s and so should not incur significantly more disk usage. If desired, the index can be skipped with the --skip-index option. Note that this will make most all downstream commands much slower and filtering cannot be applited without this index file.
Additional Command Line Options¶
--num-most-common-errors
Dynamically updates the most common reasons for a read to be unsuccessfully processed. (May cause issues for smaller viewing screens or certain environments, so this is off by default)
--failed-reads-filename
This option outputs the filenames for each read that failed via each failure mode. This can be useful for tracking down bothersome errors.
--signal-matching-score, --q-score, --obs-per-base-filter
Filter reads based on the specified criterion and threshold. These filters are applied only to the Tombo index file and can be cleared later. The
--q-scoreand--obs-per-base-filteroptions are off by default and the--signal-matching-scoredefault threshold is listed in the command help output (this value has been optimized for DNA and RNA data types). See the Read Filtering Commands section for more details.
--ignore-read-locks
Multiple independent
tombo resquigglecommands on the same set of reads should NOT be run simultaneously. This can cause hard to track errors and read file corruption. To protect against this, Tombo adds a lock (only acknowledged by Tombo) to each directory being processed. If a previoustombo resquigglecommand fails in a very unexpected fashion these locks can be left in place. In this case the--ignore-read-locksoption is provided. This is the only intended use for this option.
Pre-process Raw Reads¶
Nanopore raw signal-space data consumes more memory than sequence-space data. As such, many users will produce only FASTQ basecalls initially from a set of raw reads in FAST5 format. The Tombo framework requires the linking of these basecalls with the raw signal-space data. The annotate_raw_with_fastqs sub-command is provided to assist with this workflow.
Given a directory (or nested directories) of FAST5 raw read files and a set of FASTQ format basecalls from these reads, the annotate_raw_with_fastqs adds the sequence information from the FASTQ files to the appropriate FAST5 files. This command generally adds 15-25% to the disk usage for the raw reads.
This functionality requires that the FASTQ seqeunce header lines begin with the read identifier from the FAST5 file. This has been tested with the Oxford Nanopore Technologies supported basecaller, albacore. Third-party basecallers may be not be processed correctly.
tombo preprocess annotate_raw_with_fastqs --fast5-basedir <fast5s-base-directory> --fastq-filenames reads.fastq
Non-standard Data Locations¶
In the Tombo framework, it is possible to access and store basecalls and genome-anchored re-squiggle results in custom locations within FAST5 files.
For example, basecalls can be found in the Basecall_1D_001 slot in a set of reads that have been basecalled more than one time. In this case the basecalls can be accessed in Tombo by specifying the --basecall-group option to the tombo resquiggle command.
It can also be adventageous to store re-squiggle results in a non-standard locations. For example, if one would like to test multiple sets of re-squiggle parameters or reference versions without having to overwrite old results and re-run the tombo resquiggle command, the --corrected-group option can be specified. This will store the re-squiggle results in a new slot within the FAST5 file as well as creating a new Tombo index file.
Important
If the --corrected-group is specified in the tombo resquiggle command, this same value must be passed to all other Tombo sub-commands in order to access these results. This inlcudes all filtering, plotting, significance testing, and text output commands.