Modified Base Detection¶
Tombo enables four methods (including two sample comparison methods) for detecting shifts in raw current signal level, indicative of non-canonical bases. These four methods allow researchers to investigate non-canonical bases given any sample type, while enabling more accurate detection of specific modifications when applicable.

Tombo modified base testing methods.¶
All four methods are accessed by the tombo detect_modifications command group as described below.
TL;DR:
Motif-specific models (new in version 1.5; E. coli dcm and dam and CpG methylation motifs available) provide the most accurate modified base detection and are thus the preferred method where applicable.
Access these models via the
tombo detect_modifications alternative_modelcommand with the--alternate-bases dam dcm CpGoption.
All-context alternate models (less accurate than motif models) identify 5-methylcytosine (5mC; DNA or RNA) and N6-methyladenosine (6mA; DNA only) in any sequence context, run
tombo detect_modifications alternative_modelwith the--alternate-bases 5mC 6mAoption.For more experimental de novo modified base detection run
tombo detect_modifications de_novoproviding only a set of reads to compare with the canonical base modelFor modified base detection via comparison to a control sample (e.g. PCR or IVT) run
tombo detect_modifications model_sample_comparewith a control set of reads (--control-fast5-basedirs)For modified base detection via comparison to any second sample run
tombo detect_modifications level_sample_comparewith a second set of reads (--alternate-fast5-basedirs)Each
tombo detect_modificationscommand will produce a binary file (not intended for use outside the Tombo framework)To extract useful text files see the
tombo text_outputcommandsTo visualize raw signal around significant regions use the
tombo plot most_significantcommandTo assess testing results given a ground truth (motif or previously identified sites) use the
tombo plot motif_with_stats,tombo plot roc,tombo plot sample_compare_roc,tombo plot per_read_rocandtombo plot sample_compare_per_read_roccommands
Hint
The tombo resquiggle command must be run on a set of reads before processing with tombo detect_modifications.
Specific Alternate Base Detection (Recommended)¶
In order to detect a specific non-canonical base, use the tombo detect_modifications alternative_model command. This command identifies sites where signal matches the expected levels for an alternate base better than the canonical expected levels. This command computes a statistic similar to a log likelihood ratio (LLR) but dynamically scaled to be more robust to outlier signal levels. This statistic is computed for each “swap base” within each read provided (e.g. each cytosine for 5mC detection or each adenine for 6mA detection).
This statistic is computed by scaling the LLR by the normal likelihood function with the same variance and mean halfway between the canonical and alternate expected signal levels. Three additional scaling factors are added to this function in order to give greater weight to sequence contexts with larger differences between the canonical and alternate expected signal levels, which inherently provide more power to distinguish the canonical and alternate base signal levels. These parameters are also set so that values are on relatively the same scale as a log likelihood ratio for setting --single-read-threshold values. Default values for the scale factors below are \(S_f = 4\), \(S_{f2} = 3\) and \(S_p = 0.3\), which produce the functions shown in the figure below. Users can experiment with the effect of these parameters with the provided scripts/outlier_robust_llr.R script.
In order to compute a standard log likelihood ratio, use the --standard-log-likelihood-ratio option.
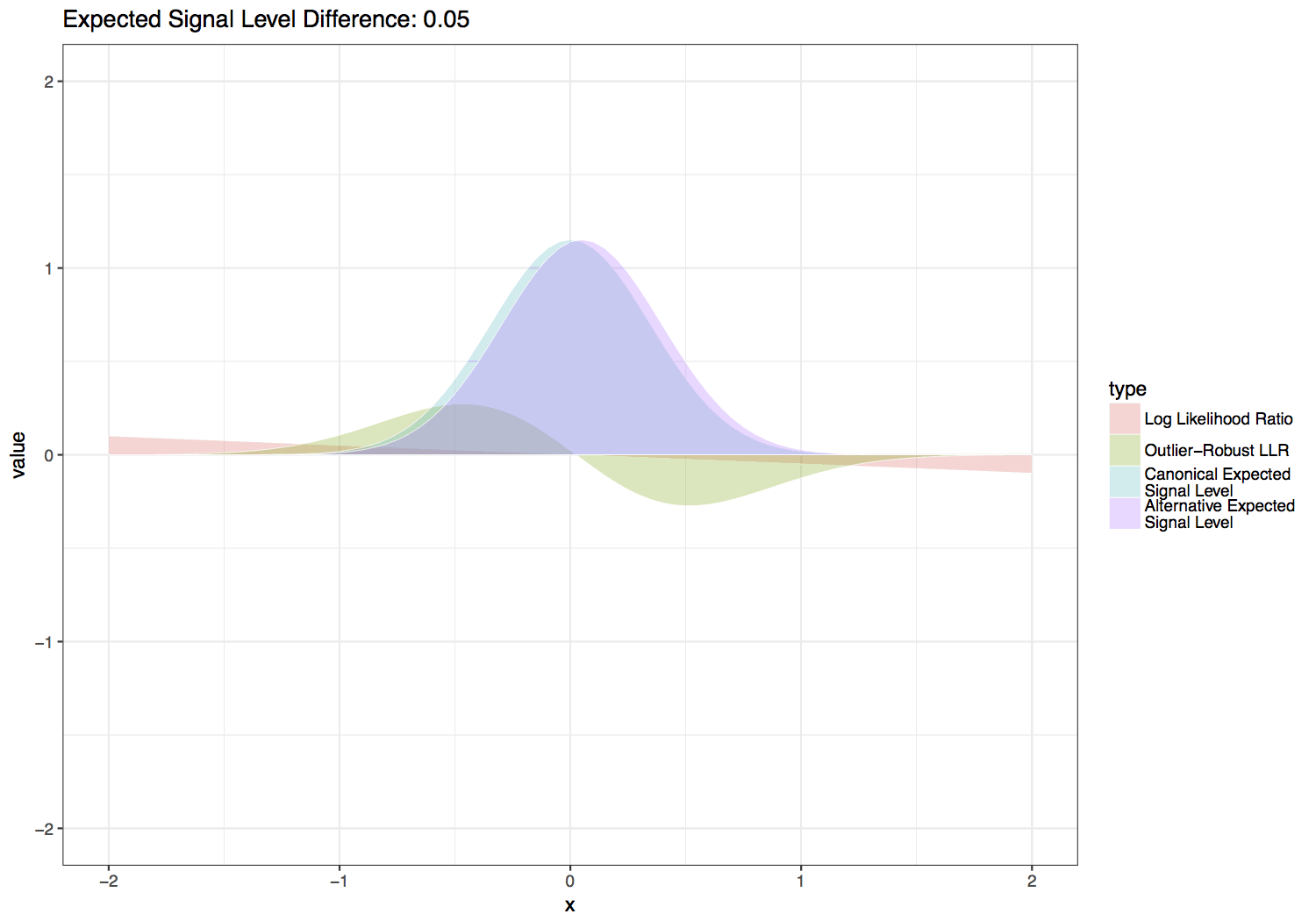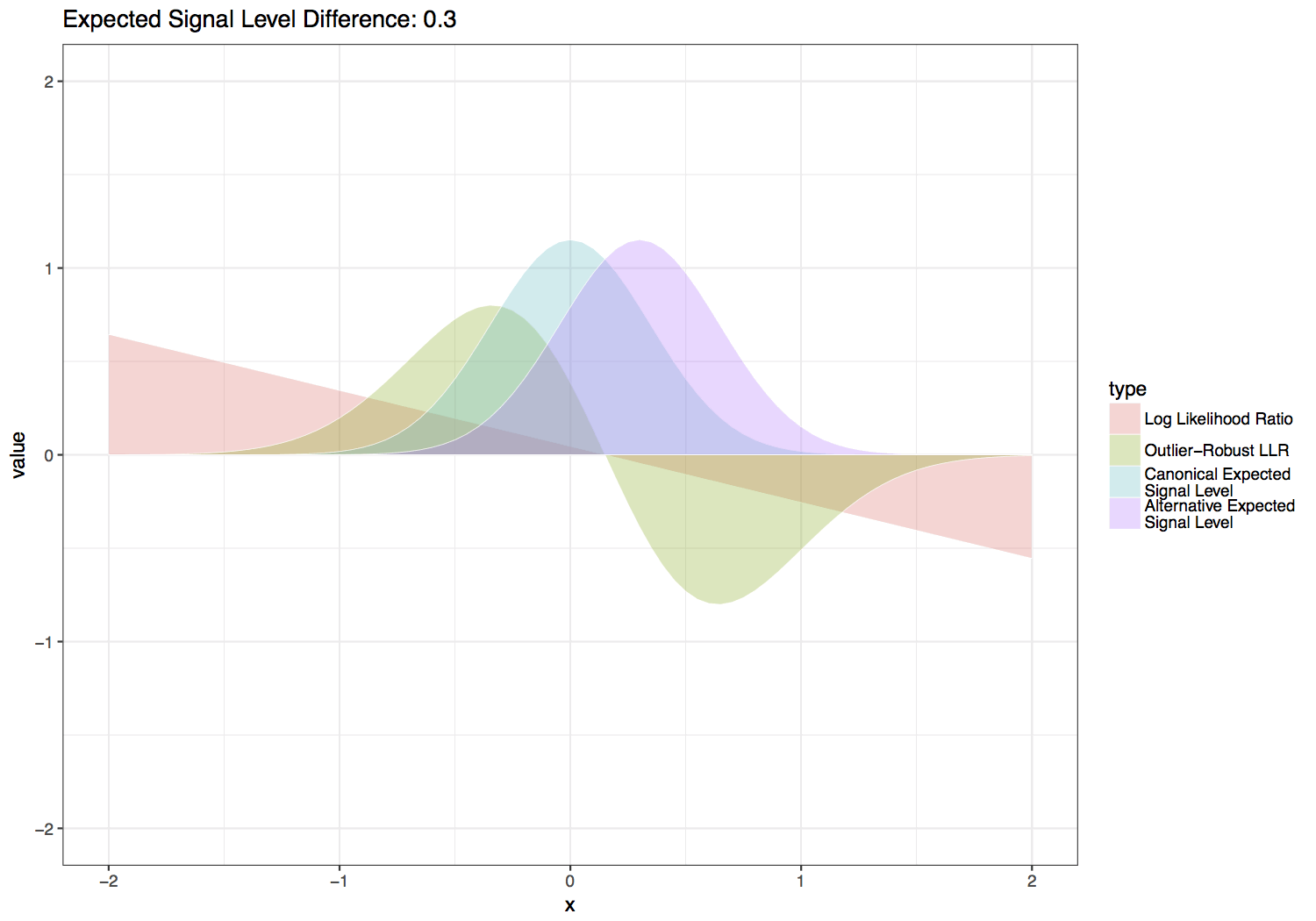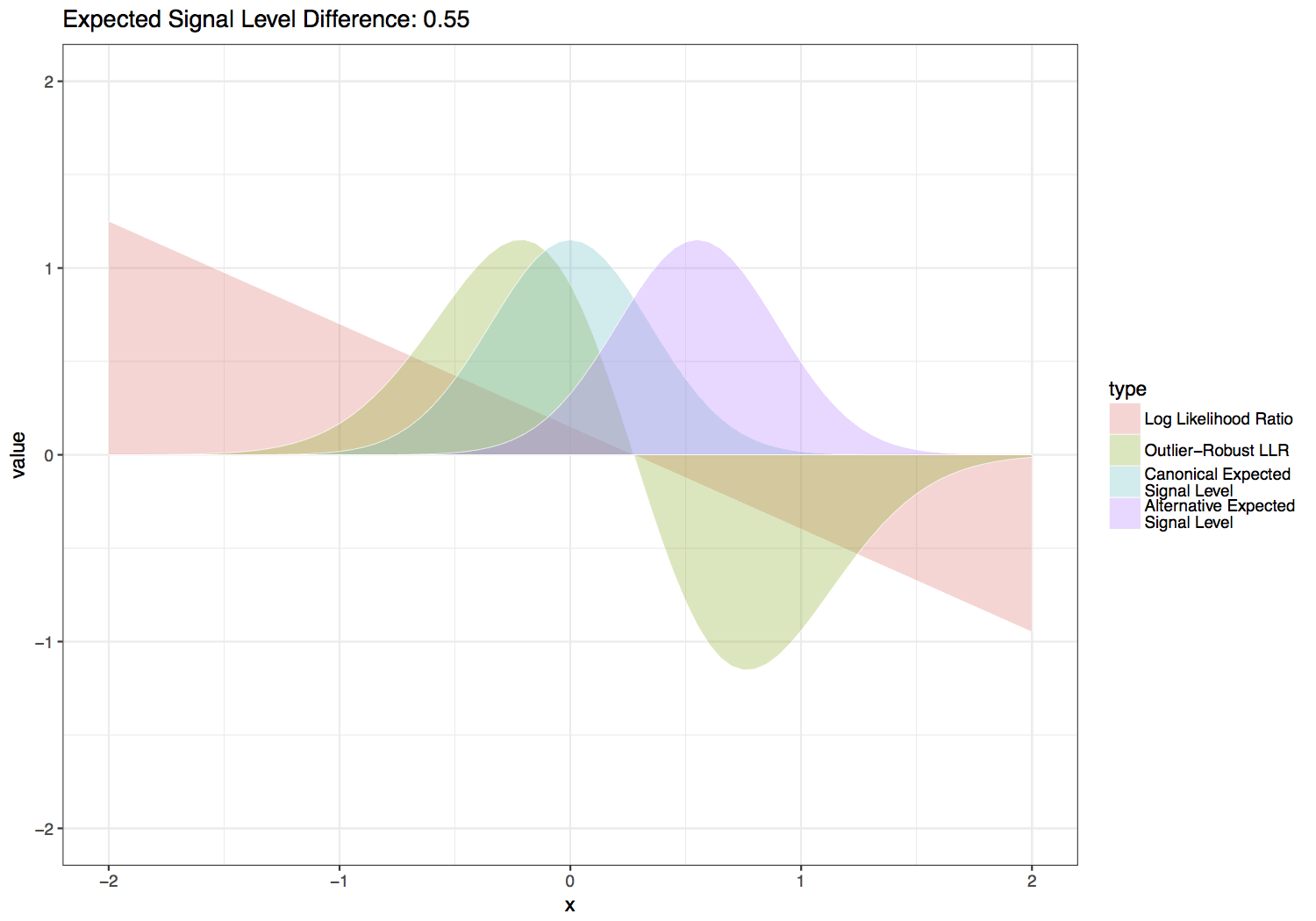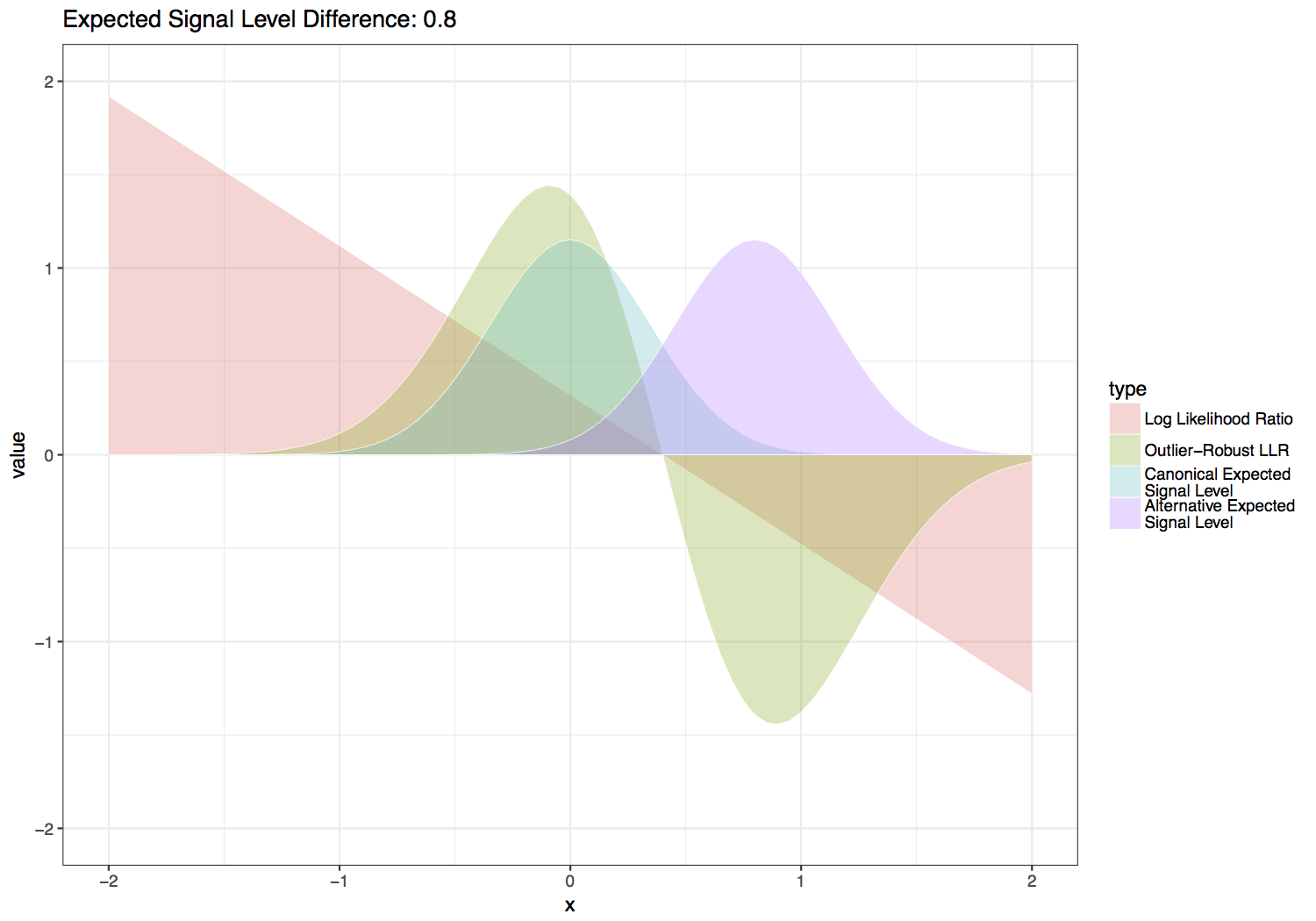
Tombo outlier-robust versus standard likelihood ratio statistic over varied differences between canonical and alternate expected signal levels.¶
This statistic is computed and summed over all positions where the base of interest is included in the modeled k-mer. The default DNA model is a 6-mer, so the signal at the six surrounding genomic bases contribute to the resulting statistic at any one position. For example, for 5mC detection within in a TGGTA C GTCCG context, the signal will be tested against expected canonical and alternate 5mC levels at the following locations:
TGGTA **C** GTCCG
-----------------
TGGTA **C**
GGTA **C** G
GTA **C** GT
TA **C** GTC
A **C** GTCC
**C** GTCCG
New alternate base models will be added as they are trained and validated internally. This is the perferred method for modified base detection if a model is available for your biological sample of interest as the exact modification position is identified.
Motif-specific models were added in version 1.5 and provide more accurate results based on the model estimation procedure. These motif-specific models will also help to improve the all-context models in the future. Motif-specific models are available for E. coli dam and dcm methylation as well as CpG methylation.
Motif-specific models are much more user-friendly to train as seen in the Model Training (Advanced Users Only) section.
tombo detect_modifications alternative_model --fast5-basedirs <fast5s-base-directory> \
--alternate-bases CpG --statistics-file-basename sample.alt_model
De novo Non-canonical Base Method¶
In order to perform de novo non-canonical base detection, use the tombo detect_modifications de_novo command. This method is ideal for unknown modification motif detection when using in combination with the tombo text_output signif_sequence_context command and motif detection software (e.g. MEME).
For each read at each position, this method performs a hypothesis test against the canonical model based on the genomic sequence. Note that this method can be quite error prone and may result in a high false positive rate. This method has the advantage of being the lowest barrier to entry, requiring only a set of reads and a reference sequence, allowing any nanopore researcher to start investigating potentially any type of modified base.
tombo detect_modifications de_novo --fast5-basedirs <fast5s-base-directory> \
--statistics-file-basename sample.de_novo
Canonical Sample Comparison Methods¶
As of version 1.5, Tombo provides two sample comparison methods for modified base detection (model_sample_compare and level_sample_compare).
The model_sample_compare method (equivalent to sample_compare method from Tombo versions <1.5) uses a control set of reads (e.g. PCR for DNA or IVT for RNA; provided via the --control-fast5-basedirs option) to adjust the canonical model for un-modeled local effects. This locally adjusted model is then used as in the de_novo method to identify deviations from this expected level. The amount of adjustment based on the observed levels can be controlled with the --model-prior-weights option, which essentially sets a number of pseudo-observations supporting the canonical model. Use the --sample-only-estimates option to estimate the local expected level only from observed reads (recommended only for high coverage samples).
The level_sample_compare method (new in version 1.5) compares two sets of reads to identify inequality in signal level distributions. This method, unlike the other three detection methods, does not perform per-read testing, but compares the two groups of signal levels at each reference position. This method applies either a KS-test (default), U-test or T-test and saves either an effect size statistic (default) or significance p-value. For each test the effect size statistics are the D-statistic for the KS-test, the common language effect size for the U-test (transformed to abs(0.5 - S) * 2 to result in a 0 to 1 scale with 1 indicating a modification) and Cohen’s D for the T-test.
It is recommended that a higher --minimum-test-reads value be set for the level_sample_compare command (default is 50) in order to obtain a reliable estimate for the effect size and avoid high false positive rates. This method can be the most reliable for many direct RNA applications where a comparison sample is available.
tombo detect_modifications model_sample_compare --fast5-basedirs <fast5s-base-directory> \
--control-fast5-basedirs <control-fast5s-base-directory> \
--statistics-file-basename sample.model_compare_sample
tombo detect_modifications level_sample_compare --fast5-basedirs <fast5s-base-directory> \
--alternate-fast5-basedirs <alternate-fast5s-base-directory> \
--statistics-file-basename sample.level_compare_sample
Note
Due to the nature of nanopore sequencing, the context surrounding the read head effects the electric current observed at any position. Thus shifts in signal due to a modified base may occur at several positions to either side of the true assigned modified base location. In order to account for this, the canonical sample and de novo modfied base detection methods accept the --fishers-method-context option which combines test values, using Fisher’s Method, over a moving window across each read. For the level_sample_compare method the statistics are averaged over this window. This can help to center significant values on true modified base positions. The default value for this parameter is 1, but reasonable results can be obtained for values between 0 and 3.
Aggregating Per-read Statistics¶
All of the above methods (except the level_sample_compare method) compute per-read, per-genome location test statistics. In order to facilitate research at the reference location level, these per-read statistics are combined at each reference position by applying a global threshold identifying each read as supporting a canonical or alternate base. This results in a fraction of reads indicating a modified base at each reference position. This global threshold may consist of a single threshold value or a pair of values (where test statistics between the values do not contribute to the estimated fraction of modified reads).
All tombo detect_modifications methods enable output of per-read test statistics (--per-read-statistics-basename). Tombo also provides the tombo detect_modifications aggregate_per_read_stats command in order to apply different global threshold values to per-read statistics without re-computing these statistics. Note it is not possible to change other testing parameters from this command (e.g. --fishers-method-context).
Dampened Fraction Estimates¶
At low coverage locations the fraction of modified reads estimates can be poor. Thus the --coverage-dampen-counts option is provided in order to dampen the estimated fraction of modified reads at low coverage locations. This allows easier use of the dampened fraction statistic in downstream analysis.
The fraction estimate includes pseudo-counts added to the un-modified and modified read counts (as specified by the
--coverage-dampen-countsoption)This is equivalent to using a beta prior when estimating the fraction of reads modified at each position
Test the effect of different dampen counts using the
scripts/test_beta_priors.R(the default values are shown below)The raw fraction is still included in the statistics file as well

Heatmap showing the resulting dampened farction of modified reads given the default --coverage-dampen-counts values over range of coverage and number of un-modified reads for default --coverage-dampen-counts 2 0.¶
Multi-processing¶
Tombo statistical testing provides the option to perform testing spread across multiple processes. This also limits the memory requirement for modified base detection, as only signal levels within a multiprocess block are held in memory. For very high coverage samples, consider lowering the --multiprocess-region-size value to minimize computational memory usage.
Multi-processing is performed over batches delineated by regular intervals across chromosomes covered by at least one read. The interval size is determined by the --multiprocess-region-size option and processed by a number of processors indicated by the --processes option. The produced per-base (and per-reda) results are identical no matter the multi-processing options selected. These regions are also used as batches to store the pre-read statistics file.
Tombo Statistics File Format¶
For all modified base detection methods, the result is a binary Tombo statistics file. This file contains statistics associated with each genomic base producing a valid result. This file is not intended for use outside of the Tombo framework. Several Tombo commands (e.g. tombo text_output browser_files, tombo text_output signif_sequence_context and tombo plot most_significant) take the binary statistics file as an input, accommodating many user pipelines downstream of modified base detection.
While the Tombo statistics file is meant to be a binary file not processed by outside tools its contents are described here for completeness. Access to this file is recommended through the tombo.tombo_helper.TomboStats object in the Tombo python API.
The Tombo statistics file is in HDF5 format. Attributes at the root level are 1) stat_type indicating which testing method was used (model_compare, de_novo, model_sample_compare, or level_sample_compare), 2) block_size indicating the number of genomic bases in each statistics block and 3) Cov_Threshold` containing the coverage threshold applied to this file (except for level_sample_compare files).
Blocks of statistics are stored in the Statistic_Blocks group. Within this group, each block of statistics is found within a group named Group_NNN. Each group contains attributes for the block start, chrm and strand. The block_stats data set contains the per-location statistics records. Each record contains the following attributes: damp_frac, frac, pos, chrm, strand, cov, control_cov, and valid_cov. For level_sample_compare files the damp_frac and frac values are replaced by the stat value.
frac contains the fraction of valid (not including per-read statistics within the interval specified by --single_read_threshold) reads at this genomic position identified as the standard base.
cov, control_cov, and valid_cov contain the read coverage at the genomic position for the sample and control reads. control_cov is only applicable for the control sample comparison testing method. valid_cov contains the number of reads contributing to the frac of tested reads as defined by --single-read-threshold.
Per-read Statistics File Format¶
Per-read statistics can be stored by setting the --per-read-statistics-basename option to any tombo detect_modifications command. This output file can then be used in downstream Tombo sub-commands (e.g. the tombo plot per_read and tombo detect_modifications aggregate_per_read_stats commands).
For advanced users, the Tombo per-read statsitics file can be accessed via the Tombo python API using the tombo.tombo_stats.PerReadStats class. This class provides initialization, simply taking the per-read statsitics filename. The PerReadStats class supports the get_region_stats function which takes a tombo.tombo_helper.intervalData object specifying an interval of interest. This will return a numpy array containing a record for each read (specified by the read_id field) and each tested genomic position (pos field) along with the test statistic (stat field) at that location.
Important
All other optional arguments to the tombo.tombo_stats.PerReadStats constructor should be left as None; setting these values will delete the file and construct a blank per-read statistics file.
The per-read statistics file is in the HDF5 format. All blocks are stored within the Statistic_Blocks slot. The size of the blocks is stored in the block_size attribute (defined by the --multiprocess-region-size option) and the type of statistical test applied is stored in the stat_type attribute.
Each genomic block is stored in a different Block_[##] slot. These slots do not have any particular order. Within each block the chrm, strand and start of the block are stored. The block statistics are stored in the block_stats slot. Per-read statistics contain a record for each tested location within each read. Each record contains the genomic position (pos), the test statistic (stat; hypothesis test p-value or log likelihood ratio as indicated by the statistic type), and the read_id. A single read spanning multiple blocks will contain statistics in more than one block. An individual read’s statistics can be reconstructed using the read_id field.